





P-592 - CARCINOMA DE MAMA METAPLÁSICO: A PROPÓSITO DE UN CASO CLÍNICO EXCEPCIONAL
1Hospital Clínico Universitario Virgen de la Victoria, Málaga; 2Hospital San Agustín, Linares.
Introducción: El carcinoma metaplásico de mama es una forma poco común y agresiva de cáncer mamario. Su diagnóstico es complejo y su tratamiento desafiante. Se presenta a continuación un caso de carcinoma metaplásico de una paciente intervenida en nuestro centro.
Caso clínico: Paciente de 45 años, fumadora activa, intervenida de cesárea y mamoplastia de aumento. Consulta por masa dolorosa en cuadrante superoexterno de mama derecha. A la exploración se palpa tumor adherido a piel y planos profundos (prótesis). No se palpan adenopatías axilares. En la mamografía: nódulo solidoquístico de 85 mm, palpable, de rápido crecimiento. La biopsia confirma una neoplasia maligna pobremente diferenciada, sin expresión de receptores hormonales, HER2 ni marcadores epiteliales. Se decide en comité multidisciplinar mastectomía radical con exéresis de prótesis y el complejo areola-pezón. El estudio anatomopatológico revela carcinoma metaplásico invasor de tipo sarcomatoide (99%), con componente epitelial (1%) y ductal in situ de alto grado con necrosis (< 5%). El tumor mide 10 cm, infiltra músculo, y es grado III (índice Ki-67 del 90%). Estadio pT4aNxM0 (IIIB). Inicia tratamiento adyuvante con quimioterapia para cáncer triple negativo. Evoluciona clínicamente bien, sin evidencia actual de enfermedad.
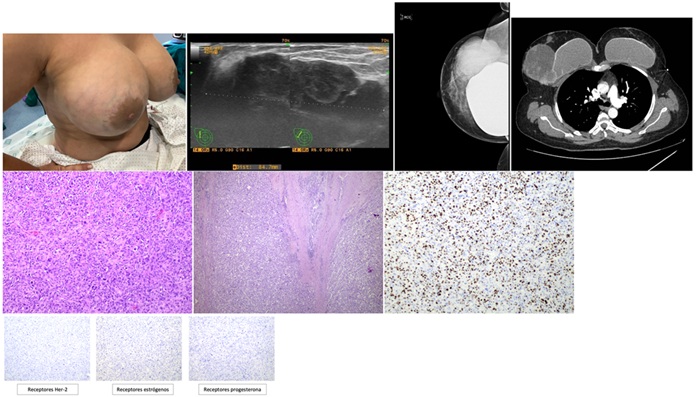
Discusión: El carcinoma metaplásico de mama (CMM) es un subtipo raro y agresivo de cáncer mamario, que representa menos del 1% de los casos. Se caracteriza por su heterogeneidad morfológica, con componentes epiteliales y mesenquimales que incluyen diferenciación escamosa, sarcomatoide o condro-ósea. Suele presentarse en mujeres entre los 50 y 60 años y, a diferencia de otros tipos de cáncer de mama, en más del 90% de los casos tiene un perfil triple negativo (sin expresión de receptores de estrógeno, progesterona ni HER2), lo que limita las opciones terapéuticas dirigidas. Clínicamente, se manifiesta como una masa palpable de crecimiento rápido, que a menudo es de gran tamaño al momento del diagnóstico. Aunque tiene una baja tasa de afectación ganglionar, presenta una alta propensión a la diseminación hematógena, especialmente a pulmones, huesos y cerebro. El diagnóstico definitivo requiere estudios histopatológicos e inmunohistoquímicos complejos, dada su diversidad celular. El tratamiento incluye cirugía como pilar principal, combinada con quimioterapia, aunque esta última muestra limitada eficacia. La radioterapia puede ser útil en algunos casos. Debido a su comportamiento agresivo y mal pronóstico, se recomienda un enfoque multidisciplinario en centros especializados.

