




Wilson's Disease (WD) is an autosomal recessive disorder of copper metabolism resulting in a pathological accumulation of this metal, initially in the liver and later in other organs, mainly brain. Treatment with copper chelating agents and zinc salts results in a depletion of copper deposits and prevents or reverses the clinical manifestations. Copper deficiency may cause haematological and neurological changes, the latter principally being polyneuropathy and myelopathy. We report a patient with WD who developed a myelopathy associated with a deficiency of copper following prolonged treatment with D-penicillamine and zinc salts.
La enfermedad de Wilson (EW) es una enfermedad autosómica recesiva del metabolismo del cobre que provoca la acumulación patológica de este metal, primero en el hígado y posteriormente en otros órganos, principalmente el cerebro. El tratamiento con agentes quelantes del cobre y sales de zinc conduce al agotamiento de los depósitos de cobre y previene o revierte las manifestaciones clínicas de esta enfermedad. El déficit de cobre puede causar alteraciones hematológicas y neurológicas, entre estas últimas principalmente polineuropatía y mielopatía. Se presenta un paciente con EW que ha desarrollado una mielopatía asociada con la deficiencia de cobre tras un tratamiento prolongado con D-penicilamina y sales de zinc.
Wilson's Disease is an uncommon autosomal recessive condition caused by the pathological accumulation of copper in tissues1,2. The affected gene, ATP7B, encodes a copper transport protein which is principally expressed in the hepatocyte and is necessary for the biliary excretion of this metal. The gene mutation results in dysfunction of this protein, causing excessive deposition of copper, primarily in the liver and later in other tissues, mainly brain1,2. The liver abnormalities may be variable, from asymptomatic elevation of transaminases to acute liver failure or decompensated cirrhosis1,2. The fall in serum levels of ceruloplasmin is due to a failure of incorporation of copper into this protein, resulting in reduced half-life. The neurological manifestations attributable to this condition are principally Parkinsonian features, behavioural abnormalities, mood disorders and psychotic symptoms3.
The pharmacological agent of greatest experience in the treatment of WD is D-penicillamine, a copper chelating agent which promotes its renal excretion. Its efficacy has been demonstrated and it is the therapeutic agent of choice, especially in symptomatic cases with liver manifestations1,2. In patients with asymptomatic liver abnormalities, as well as in symptomatic patients after initial chelation with D-penicillamine, zinc salts are recommended because of their greater tolerance1,2,4,5. Zinc induces the synthesis of metallothionein in the intestinal mucosa, an endogenous copper chelating agent which binds to it and prevents its absorption. Copper present in saliva and gastric juice is chelated and this leads to an increase in faecal excretion, creating a negative balance and thereby depleting stored copper6.
Copper is necessary for the proper functioning of various important enzymes such as cytochrome-c oxidase in the mitochondrial respiratory chain, zinc-copper superoxide dismutase in antioxidant mechanisms and dopamine monooxygenase in catecholamine synthesis, amongst others7. Deficiency leads to haematological abnormalities, mainly sideroblastic anaemia, neutropaenia and even to a picture resembling myelodysplastic syndrome8. It may also cause neurological changes such as polyneuropathy and a myelopathy similar to subacute combined degeneration due to vitamin B12 deficiency9–11.
We describe a patient with WD undergoing long-term treatment, first with D-penicillamine and later with zinc, who developed a myelopathy due to copper deficiency.
Clinical caseA 56 year old female had been diagnosed with advanced WD at 18 years old following her presentation with cirrhosis and bleeding oesophageal varices. She was found to have reduced serum ceruloplasmin levels, raised urinary copper, Kayser-Fleisher rings and histologically confirmed cirrhosis with hepatocytes staining positive for copper. ATP7B gene studies showed that she was homozygous for the H1069Q mutation. Because of further episodes of variceal haemorrhage, she first had a splenorenal shunt and, some years later, oesophagogastric devascularisation, oesophageal transection and splenectomy. Ten years later, she developed a portomesenteric thrombosis.
Since the time of diagnosis of WD, she was treated with low-copper diet and D-penicillamine, 750mg daily. Measurement of urinary copper during the last ten years of follow-up ranged from 200 to 500μg daily and her treatment was changed to zinc, 150mg daily (equivalent to 503mg zinc acetate), three years ago, reducing the amount of urinary copper to 40μg daily with a ceruloplasmin level of 14mg/dl. As regards her cirrhosis, neither ascites nor hepatic encephalopathy had ever been a feature and she now has only small oesophageal varices as well as a mild hypertensive gastropathy.
She recently presented with symptoms of slowly progressive unstable gait of six months duration associated with paraesthesia of the hands and feet. Neurological examination showed generalised hyperreflexia, bilateral plantar flexion reflexes, hypoaesthesia in the palms and feet, absent vibratory sensation in the feet, ataxic gait and a positive Romberg's sign.
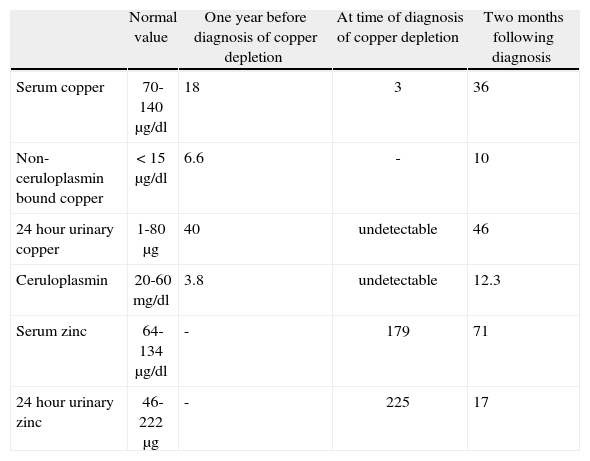
Further analyses showed the following: haemoglobin 11.3g/dl, MCV 109 fl, leucocytes 3100/mm3 (neutrophils 700/mm3, remainder normal), platelets, sedimentation rate and coagulation screen normal, ALT 38 UI/l, AST 68 UI/l, alkaline phosphatase 373 UI/l, γ-glutamyltransferase 32 UI/l, bilirubin normal. Ceruloplasmin was undetectable (normal 20-60mg/dl), serum copper 3μg/dl (normal 70-140μg/dl), 24hour urinary copper undetectable (normal 1-80μg/24h), serum zinc 179μg/dl (normal 64-134μg/dl), 24hour urinary zinc 225μg (normal 46-222μg) (table 1) and urinary lead undetectable.
Laboratory parameters.
| Normal value | One year before diagnosis of copper depletion | At time of diagnosis of copper depletion | Two months following diagnosis | |
| Serum copper | 70-140μg/dl | 18 | 3 | 36 |
| Non-ceruloplasmin bound copper | < 15μg/dl | 6.6 | - | 10 |
| 24hour urinary copper | 1-80μg | 40 | undetectable | 46 |
| Ceruloplasmin | 20-60mg/dl | 3.8 | undetectable | 12.3 |
| Serum zinc | 64-134μg/dl | - | 179 | 71 |
| 24hour urinary zinc | 46-222μg | - | 225 | 17 |
The following analyses were either normal or unremarkable: glucose, creatinine, albumin, cholesterol, triglycerides, protein electrophoresis, immunoglobulins, TSH, free T4, parathyroid hormone, Fe, ferritin, vitamin B12, folate, autoantibodies (ANA, antiDNA, ENA and ANCA), serology for HBV, HCV, HIV, syphilis and Lyme Disease.
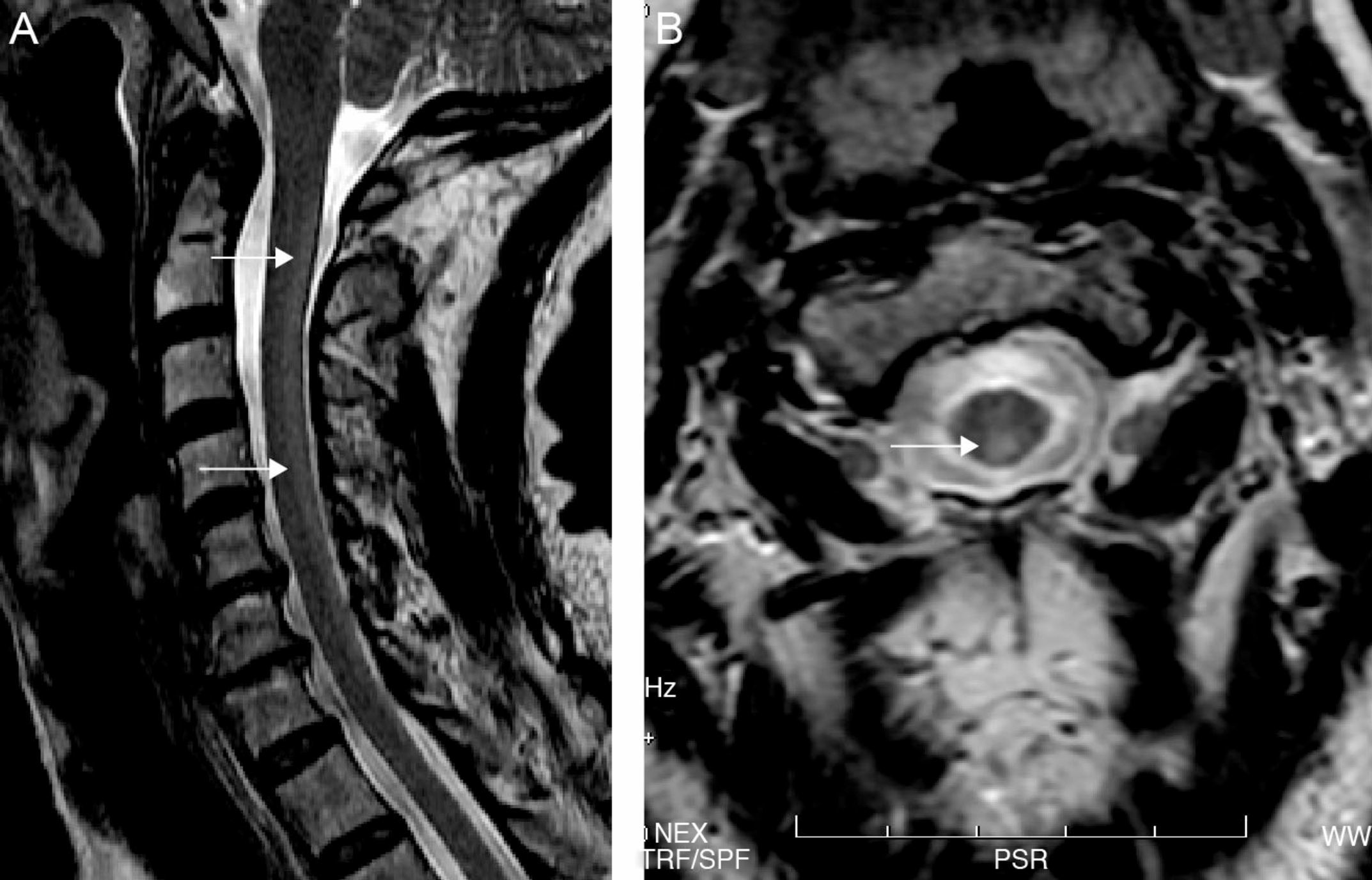
Both electroneurography and electromyography studies were normal and MRI showed hyperintense T2-weighted imaging in the posterior half of the cervical cord from C2 to C7 and also in the periventricular white matter (Fig. 1). No abnormalities were found in deep grey nuclei. Somatosensory evoked potentials from the median nerve were normal but those from the posterior tibial nerve were seen to elicit responses in the lumbar region but without cortical responses, findings suggestive of involvement of the posterior columns.
 and axial (B) T2-weighted cervical MRI. Hyperintense image in the posterior half of the cervical cord from C2-C7 (arrows).")
Treatment with zinc was discontinued and oral copper was commenced in order to achieve a normal non-ceruloplasmin bound copper and urinary copper excretion (table 1). Zinc was later recommenced at a lower dose. The patient's symptoms improved minimally over the first few months and later stabilized.
DiscussionWe have presented a case of WD which was diagnosed on the basis of histological cirrhosis with copper granules on liver biopsy, low ceruloplasmin, raised urinary copper excretion, Kayser-Fleisher rings and with consistent genetic studies. She received chelation therapy with D-penicillamine for 35 years and this was later changed to zinc acetate for three years. During follow-up, urinary copper levels suggested good compliance with treatment and, in that time, no deterioration in liver function or neurological symptoms was noted. During the third year of treatment with zinc, she presented with an unsteady gait due to sensory ataxia. A chronic hepatic encephalopathy was initially ruled out due to normal mental status and absence of extrapiramidal alterations or sleep disturbances, characteristic of this condition and that differ from the clinical picture of our patient. Somatosensory evoked potentials detected alterations in the spinal cord proprioceptive pathways and MRI showed changes consistent with myelopathy. Vitamin B12 levels were normal and copper deficiency was detected.
Although there is an excess of copper in WD, serum levels are usually low and non-ceruloplasmin bound copper is a better test than is total serum copper1,2. It can be calculated as the difference between the concentration of serum copper (in μg/dl) and a 3-fold concentration of ceruloplasmin (inmg/dl). Its normal value is lower than 15μg/dl and in many patients with WD levels of greater than 25μg/dl are found1,2. Non-ceruloplasmin bound copper has great value as a measure of response to treatment12, but it is also useful in detecting copper depletion which may occur when the level falls to less than 5μg/dl1. In the case of treatment with D-penicillamine, low values of urinary copper may be an indication of overtreatment, then accompanied by very low values of non-ceruloplasmin bound copper1. In the case of treatment with zinc, one of the aims is to achieve a urinary copper of less than 75μg/24hours and, for this reason, unlike D-penicillamine, low levels per se do not suggest overtreatment1, unless they are extremely low, as in our patient. In patients with excessive depletion of copper, other abnormalities may appear including lowering of ceruloplasmin levels, anaemia and neutropaenia8–11.
Our patient had a deficiency of copper as shown by a very low level of serum copper and undetectable ceruloplasmin and urinary copper and, undoubtedly, non-ceruloplasmin bound copper was also very low considering the total copper value measured. The anaemia and neutropaenia detected were almost certainly due to portal hypertension since these figures remained stable during follow-up and, indeed, in published series, haematological manifestations are not always associated with neurological features9–11.
Cases of copper deficiency in various clinical situations have been reported, including those following gastrectomy or gastric bypass surgery9–11, intestinal malabsorption syndromes such as coeliac disease13 and secondary to excessive ingestion of zinc as well as medication or denture adhesives10,11,14. Zinc overload leads to a deficiency of copper and it is this which seems to be responsible for all the clinical manifestations.
Although it appears paradoxical, copper deficiency in WD has been documented. In some series, patients have been described with a suspicion of deficiency although no exact details were provided5,15. Four cases of WD have been described with neurological abnormalities attributed to copper deficiency. They had received treatment for between five and 28 years, all with zinc salts and, in three of them, this was coprescribed with a chelating agent (D-penicillamine or trientine). In two cases16,17 a polyneuropathy was described and a myelopathy was suspected because of alterations in somatosensory evoked potentials, although unlike our case, spinal cord MRI was normal in both. Another patient presented with status epilepticus18 and a further showed cerebral demyelination but this latter case was open to question because non-ceruloplasmin bound copper was not low and urinary copper levels were not provided19. Our patient is only the second reported case of WD presenting with copper deficiency during monotherapy with zinc and, as with those cases of aforementioned myelopathy secondary to copper deficiency11, the neurological sequelae persisted during the period of follow-up provided.
In conclusion, although copper deficiency in patients with WD is very infrequent and may not be obvious, it is important to remember this possibility and to bear in mind the need for close clinical and biochemical monitoring throughout treatment.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

