






INTRODUCTION
Abnormalities of cardiopulmonary function are present in most patients with advanced liver disease1. A combination of pulmonary and hepatic risk factors can lead to the synchronous existence of liver and lung disease, as seen in combined alcohol, drug, and nicotine consumption, often with additive negative effects. The pulmonary manifestations of liver disease include hypoxia related to tense ascites which creates a hindrance to respiratory diaphragmatic excursions, hepatic hydrothorax with compressive atelectasis, and aspiration pneumonia secondary to altered consciousness in patients with hepatic encephalopathy or ethanol abuse2 (table I).

Besides these well-recognized clinical entities, two distinct pulmonary syndromes deserve special attention when dealing with patients with advanced chronic liver disease: portopulmonary hypertension and hepatopulmonary syndrome. Both syndromes seem to be pathogenetically closely related to the presence of portal hypertension, but their pathophysiological mechanisms, still unclarified, clearly differ.
The knowledge that a subset of patients with hepatic disease develops significant pulmonary vascular alterations, either microvascular dilation leading to the hepatopulmonary syndrome or arteriolar vasoconstriction leading to portopulmonary hypertension, indicates that unique changes in the pulmonary vascular district may occur. These pulmonary vascular syndromes significantly impact morbidity and mortality in affected patients and may influence candidacy for liver transplantation3. Thus, their recognition is of paramount relevance.
The development of the portopulmonary hypertension or the hepatopulmonary syndrome has major clinical and prognostic implications and requires specific treatment considerations.
PORTOPULMONARY HYPERTENSION
The association between pulmonary artery hypertension and portal hypertension has been termed portopulmonary hypertension. From a hemodynamic standpoint it is defined by the following features: 1) mean pulmonary artery pressure > 25 mmHg at rest and > 30 mmHg on exercise, 2) increased pulmonary vascular resistance of > 120 dyne/sec/cm5, and 3) pulmonary capillary wedged pressure < 15 mmHg. According to the World Health Organisation classification, portopulmonary hypertension is no longer classified as secondary pulmonary hypertension but as a pulmonary arterial hypertension associated with liver disease or portal hypertension of either presinusoidal or postsinusoidal origin4.
An association between pulmonary hypertension and cirrhosis was first recognized in 1951 by Mantz and Graig5, and for many years it was supposed that surgical portosystemic shunting contributed to increase the risk of developing pulmonary hypertension. After this initial case report, the relationship between cirrhosis and pulmonary hypertension was confirmed in a large autopsy series where a 0.73% prevalence of pulmonary hypertension was observed in cirrhotic patients compared to prevalence of 0.13% in subjects without chronic liver disease6. By use of various diagnostic criteria (see below), 2-10% of cirrhotic patients have been estimated in clinical studies to be at risk of developing pulmonary hypertension7-10. In selected patients who have advanced liver disease, especially those assessed or referred for liver transplantation, pulmonary hypertension occurs in up to 16%11,12. Although these observations seem to suggest that portopulmonary hypertension is a complication affecting mainly patients with advanced liver disease, no clear relation between the severity of hepatic dysfunction or raised portal pressure and the severity of pulmonary hypertension has been conclusively shown.
Pathogenesis
The development of portopulmonary hypertension seems to be independent of the cause of portal hypertension. Although most patients with portopulmonary hypertension have cirrhosis, the syndrome has been described in patients with portal hypertension due to non-hepatic causes, such as portal venous disease. Thus, portal hypertension seems to be a unavoidable pre-requisite for the development of pulmonary hypertension.
The mechanism by which portal hypertension causes pulmonary hypertension remains incompletely understood. The pathogenesis is complex and involves an inter play between haemodynamic and local factors. The mechanisms proposed for the development of portopulmonary hypertension include excess local production of vasoconstrictor substances, increased pulmonary blood flow leading to endothelial damage and vascular remodelling, excess pulmonary vascular volume, and in situ microthrombosis. In human and model systems, altered local production of vasoconstrictor such as endothelin-1 and serotonin and impaired production of vasodilators such as nitric oxide and prostacyclin may play a role. Of all the vasoconstrictive factors implicated in the pathogenesis of pulmonary hypertension, endothelin-1 is the most potent. Indeed systemic endothelin-1 levels have been shown to be increased in patients with primary pulmonary hypertension. Previous studies have also demonstrated higher systemic endothelin-1 levels in cirrhotic patients compared with controls.
A hyperdynamic circulation seems to be present in almost all patients who have portopulmonary hypertension in the early stage of this condition. However, high cardiac output and hyperdynamic circulation are typical hemodynamic features in cirrhotic patients who have developed portal hypertension. In turn high cardiac output produces increased vascular shear stress on the pulmonary circulation: maintenance of an adequate pulmonary vascular resistance would prevent development of the syndrome whereas an abnormal pulmonary vascular dilation would lead to an abnormally decreased vascular resistance and hepatopulmonary syndrome. In contrast, increased vascular resistance caused by vasoconstriction and progressive pulmonary vascular remodelling due to proliferation of pulmonary arterial endothelial cells and smooth-muscles cells will result in pulmonary hypertension. The presence of a high cardiac output can result in a mild degree of pulmonary hypertension in the presence of a normal or near-normal pulmonary vascular resistance.
Portopulmonary hypertension remains of mild to moderate severity in most patients, and vasoconstriction and medial hypertrophy of the pulmonary arteries are the dominant features of pulmonary hypertension13,14. The pulmonary histological abnormalities in portopulmonary hypertension are identical to those found in primary pulmonary hypertension and includes smooth muscle proliferation and hypertrophy, concentric intimal fibrosis, plexogenic arteriopathy and necrotizing vasculitis. Obstructive intimal thickening and formation of plexiform lesions are prominently seen in blood vessels, especially at the level of small arteries and arterioles13,15. Plexiform lesions are the characteristic glomeruloid structures seen in pulmonary hypertension and result from proliferation of phenotypically distinct endothelial cells16. These lesions are located in the small distal branches of the pulmonary artery, usually at more proximal sites than occlusive lesions16-18. Neoangiogenesis may be present, as well as thromboembolic lesions19. The factors involved in the development of proliferative vasculopathy have yet not been identified.
Clinical features
The mean age of presentation of portopulmonary hypertension is the fifth decade; the distribution is the same in the two sexes. The most common symptoms and clinical signs in patients with portopulmonary hypertension are: progressive dyspnoea, fatigue and peripheral edema. Other symptoms such as palpitations, syncope or chest pain are less frequent. The severity of these symptoms worsens with increasing pulmonary hypertension20. Physical findings indicating pulmonary hypertension are generally subtle and may be completely absent. The most common findings are an accentuated pulmonary component of the second heart sound and a systolic murmur, indicating tricuspid regurgitation.
The prevalence and severity of portopulmonary hypertension does not appear to correlate with the degree of hepatic synthetic dysfunction, degree of portocollateral circulation as assessed by the measurement of the azygos blood flow or the severity of portal hypertension. However survival in pulmonary hypertension correlates with the severity of right sided heart dysfunction as assessed by the degree of elevation in the right atrial pressure and the degree of decline in the cardiac output.
Diagnosis
The diagnosis of portopulmonary hypertension requires raised pulmonary arterial pressure and resistance combined with the exclusion of other causes of pulmonary hypertension.
Although symptoms are common in portopulmonary hypertension and correlate with the severity of disease, they are non-specific. Therefore, making the diagnosis requires a high index of suspicion. Studies that have evaluated the diagnostic utility of various predictors of portopulmonary hypertension, including systemic hypertension, echocardiographic measures of pulmonary artery pressure and right ventricular dilation and chest radiographic abnormalities, are in general specific but poorly sensitive. In most cases, echocardiography provides the first and accurate indexes of suspicion leading to the diagnosis. If uncertainty remains, pulmonary hypertension can be conclusively shown, or ruled out, by right heart catheterisation. A recent study by Colle et al21 designed to compare Doppler echocardiography and right heart catheterisation in the diagnosis of portopulmonary hypertension, confirms that echocardiography is an excellent screening test and identified all patients with portopulmonary hypertension. However, this study has also shown that this non-invasive technique has one main limitation: Doppler echocardiography is not specific, because is unable to differentiate those patients that have increased pulmonary resistance (such as that observed in those subjects showing evidence of portopulmonary hypertension) from those who have high pulmonary artery pressure with normal pulmonary vascular resistance (such as that observed in hyperkinetic states and/or left ventricular dysfunction). As a result, right heart catheterisation seems to be the only reliable technique to discriminate between patients with portopulmonary hypertension and those with elevated pulmonary artery pressure.
When the diagnosis of portopulmonary hypertension has been established, differentiation between mild, moderate, and severe pulmonary arterial hypertension is helpful for prognostic and treatment considerations. There is no universally accepted definition on which this distinction is routinely based, but for practical use cardiac output is better suited for classification than the magnitude of pulmonary arterial pressure. In mild to moderate portopulmonary hypertension, normal or high cardiac output is present and the pulmonary vascular resistance is only slightly raised. However, in severe portopulmonary hypertension cardiac output is reduced and pulmonary vascular resistance is strikingly raised (table II).

Therapy
There are no long-term studies or guidelines on the use of pharmacotherapy in portopulmonary hypertension. Medical treatment for portopulmonary hypertension is palliative and is largely based on experience in primary pulmonary hypertension. Patients who have mild portopulmonary hypertension frequently have no symptoms and signs of pulmonary vascular disease. In these patients, specific treatment is not generally required; however, regular follow up examinations are advisable to monitor the potential progression of pulmonary disease. Severe portopulmonary hypertension has a poor prognosis10,22. The proposed available specific treatments are burdensome, expensive and risky and are described below.
Anticoagulation
Anticoagulation is recommended in patients with primary pulmonary hypertension because it can slow down disease progression23. Efficacy and safety of oral anticoagulants in patients with portopulmonary hypertension have not yet been assessed in clinical trials. Many patients do not receive anticoagulation because of an increased risk of hemorrhagic complications. In addition, the failure of liver synthetic function leads to low serum concentrations of coagulation factors with a global impairment of the coagulation cascade. However, according to the limited available data, there is no agreement as to whether anticoagulation should be recommended or not for patients with severe portopulmonary hypertension.
Pharmacotherapy
Due to the peculiar nature of this condition pharmacotherapy reports are scanty. In primary pulmonary hypertension, the vasomotor response to inhaled nitric oxide or intravenous epoprostenol (prostacyclin-PGI2) is currently measured and if a significant decrease in pulmonary vascular resistance and mean artery pressure is observed, then vasodilator agents are used. Administration of calcium channel blockers prolongs survival24. However, nifedipine has been reported to increase portal pressure in cirrhosis. One case report by Ribas et al25, has shown a beneficial effect of isosorbide mononitrate administration both on pulmonary and portal hypertension.
Intravenous epoprostenol is the best-studied drug in patients with portopulmonary hypertension. Epoprostenol is a potent pulmonary vasodilator which has significant antiproliferative and antiplatelet aggregating factors. It also appears to be able to reverse the remodelling of pulmonary vasculature that may be responsible for «fixed» pulmonary hypertension not responsive to vasodilators26. Due to its short-life in the pulmonary circulation (3-5 minutes), it needs to be administered continuously through an indwelling catheter. Although the efficacy and safety of epoprostenol in such patients has never been addressed in randomised controlled trials, strong evidence shows that this drug given intravenously improves hemodynamics and exercise capacity in patients with portopulmonary hypertension27-29. However, preliminary data from the Mayo Clinic suggest that intravenous epoprostenol does not improve long-term survival30. The need for continuous infusion of epoprostenol is a major drawback in the administration of this medication. Thus, continuous intravenous epoprostenol administration requires permanent central venous access and uninterrupted infusion of the drug. Other prostanoids have became available for the treatment of pulmonary hypertension; however, experience with aerosolised iloprost, oral beraprost sodium, or treprostinil in portopulmonary hypertension is still anecdotal.
The endothelin system might be a potential target in portopulmonary hypertension. The orally active selective ET-A receptor antagonist sitaxsentan and nonselective endothelial receptor blocker bosentan are being evaluated for their role in management of portopulmonary hypertension12. However, the potential liver toxicity associated with these drugs substantially limit their role in the management of portopulmonary hypertension.
Liver transplantation
The efficacy of liver transplantation as a treatment for portopulmonary hypertension also remains controversial. Based on retrospective data and clinical experience, moderate to severe portopulmonary hypertension (mean pulmonary artery pressure > 50 mmHg) is a contraindication to transplantation due to a peri-operative mortality of approximately 40% and lack of reversibility of pulmonary hypertension. Patients with mild pulmonary hypertension (mean pulmonary artery pressure < 35 mmHg) appear to have no increase in cardiopulmonary mortality after liver transplantation. The outcome after liver transplantation in intermediate severity portopulmonary hypertension (mean pulmonary artery pressure 35-50 mmHg) and in patients who have improvement in pulmonary artery pressure on medical therapy is less well defined.
After liver transplantation, congestion of the hepatic veins due to decreased right-ventricular function carries a notable risk of primary graft dysfunction. In highly selected cases, combined liver and lung transplantation or even heart-lung-liver transplantation might have to be considered31,32.
HEPATOPULMONARY SYNDROME
In patients with liver diseases the reported frequency of hepatopulmonary syndrome is between 4% to 29%33-36. This different incidence is mainly due to heterogeneity of the applied diagnostic criteria. The hepatopulmonary syndrome results from intrapulmonary microvascular dilation, and it is a well-defined cause of hypoxemia in patients who have liver diseases. Hepatopulmonary syndrome is commonly defined by the presence of hepatic dysfunction and/or portal hypertension, a widened age-corrected alveolar-arterial oxygen gradient on room air with or without hypoxemia and intrapulmonary vasodilation2. As with portopulmonary hypertension, hepatopulmonary syndrome occurs mostly in patients who have established cirrhosis and portal hypertension. The association between the severity of liver disease and the degree of hypoxemia is slight, but the risk to manifest this syndrome seems to be highest in Child C patients34,37,38. The cause and the stage of liver disease leading to portal hypertension does not seem to affect the development of the hepatopulmonary syndrome: this is evident from reports of a hepatopulmonary syndrome occurring in patients with prehepatic portal hypertension in the absence of chronic liver disease35,39, in Budd-Chiari syndrome40 and veno-occlusive disease, and even in patients with acute or chronic inflammatory liver disease (due to various etiologies) without evidence of cirrhosis or portal hypertension41-43. The association between pulmonary dysfunction and liver disease has been recognized in 1884, when a case of cirrhosis, cyanosis and clubbing of the fingers was described by Fluckinger, who termed this condition «hypoxia of cirrhosis». In later years, many new cases were reported of similar patients with chronic liver diseases who developed decreased arterial oxygen saturation because of pulmonary vascular dilatations. But the term «hepatopulmonary syndrome» was not used until 1977, when Kennedy and Knudson44 described a patient who developed dyspnoea on exertion after surgical portocaval shunting for complications of advanced alcoholic cirrhosis. More recently, studies demonstrated that as many as 40% of cirrhotics have detectable intrapulmonary vasodilation and up to 8-15% will develop impaired oxygenation leading to significant functional limitations45. However, it is now clear that the hepatopulmonary syndrome may occur in the setting of other cardiopulmonary abnormalities and may contribute significantly to gas exchange abnormalities in these patients42.
Pathogenesis
From a pathophysiological point of view, hepatopulmonary syndrome is almost exactly the opposite of portopulmonary hypertension. The mechanisms underling the development of intrapulmonary vasodilatation in hepatopulmonary syndrome have received increasing interest. Most studies in humans have challenged the concept that human hepatopulmonary syndrome is simply one feature of hepatocirculatory syndrome in cirrhosis. The aetiology of this syndrome remains unknown, however. The most commonly accepted hypothesis postulates that there is inadequate synthesis or metabolism of putative pulmonary vasoactive substances by the diseased liver, leading to a functional vasodilatation of the pulmonary vasculature ultimately producing hypoxemia46. To date, however, no particular substance has been implicated in causing these vascular dilatations, but possibilities include prostaglandins47, nitric oxide48, vasoactive intestinal peptide49, calcitonin50, glucagon, substance P51, and atrial natriuretic factor52. Experimental and clinical data suggest that increased production of nitric oxide in the lungs might play a pivotal role in the pathogenesis of hepatopulmonary syndrome. In addition, the endothelin system, especially abnormal activation and increased expression of endothelial type B receptors, has been implicated in the pathogenesis of the hepatopulmonary syndrome.
The only recognized experimental model of hepatopulmonary syndrome is the one observed in the chronic common bile duct ligation (CBDL) in the rat. These animals develop features analogues to a mild form of human hepatopulmonary syndrome and have provided a number of potentially important mechanistic insights. Anyhow fundamental questions about the relationship of this model to human disease remain.
Experimental hepatopulmonary syndrome
As reported recently by Fallon, the major authorithy in the field the pulmonary impairment in liver diseases, CBDL in the rat is the only recognized experimental model of hepatopulmonary syndrome. CBDL is distinct from human hepatopulmonary syndrome in that animals reliably develop intrapulmonary vasodilatation and gas exchange abnormalities as liver injury progresses, while only 8-15% of cirrhotic patients are affected by this syndrome. In addition, the onset of HS occurs within 2 weeks of CBDL prior to the development of histologic biliary cirrhosis and when hyperdynamic state and portal hypertension are still not advanced53. These studies suggest that CBDL may result in the development of a unique set of alterations similar to those observed in human hepatopulmonary syndrome, but do not exclude that pathogenetic mechanisms in human and experimental systems may be distinct.
Early studies focused on the role of vasoconstrictive eicosanoid production and increased pulmonary vascular permeability and peri-vascular oedema in the development of gas exchange abnormalities after CBDL54. Further studies have demonstrated that increased hepatic production and release of low levels of endothelin-1 (ET-1) is one mechanism for triggering an increase in pulmonary eNOS and vasodilatation after CBLD. This finding is supported by the evidence that low-level ET-1 infusion in animals with partial portal vein ligation, stimulates pulmonary eNOS expression and hepatopulmonary syndrome development55,56.
Recent studies have explored the role of other enzymes and mediators in the pathogenesis of experimental hepatopulmonary syndrome and have refocused interest on the accumulation of intravascular macrophages in the lung. An increase in pulmonary inducible nitric oxide synthase (iNOS) expression, in addition to an increase in eNOS expression, along with enhanced NOS activity, have been found in 6 weeks CBDL rats57. The increase in iNOS expression was observed predominately in intravascular macrophage-like cells. In further studies, treatment of CBLD animals for 6 weeks during the development of liver injury with norfloxacin abolished bacterial translocation, decreased intravascular macrophage accumulation and normalized iNOS, but not eNOS levels58. These studies support a role for bacterial translocation in the accumulation of intravascular macrophages and in the increase in pulmonary iNOS levels after CBDL and suggest that these events contribute to intrapulmonary vasodilatation.
Other substances that seems to be involved in the pathogenesis of hepatopulmonary syndrome are heme oxygenase-1 (HO-1) and carbon monoxide59. Preliminary studies have demonstrated that HO-1 expression increases progressively beginning 3 weeks after CBDL, predominately in intravascular macrophage-like cells, and show that chronic inhibition of HO activity in vivo improves, but does not normalize, vasodilatation and gas exchange abnormalities60. These findings support a role for HO-1 and carbon monoxide in the progression of intrapulmonary vasodilatation in CBDL animals. They also suggest that an interplay between nitric oxide and carbon monoxide generating systems in the pulmonary microcirculation may play a role in the progression of experimental hepatopulmonary syndrome.
Human hepatopulmonary syndrome
Hepatopulmonary syndrome has been observed in the full landscape of liver diseases. The observations by Gupta et al35 that hepatopulmonary syndrome may occur in extrahepatic portal venous obstruction, and by De et al39 of its occurrence also in hepatic venous outflow obstruction without cirrhosis, indicate that advanced impairment of liver function and cirrhosis are not necessary prerequisites for the development of hepatopulmonary syndrome. A clinical syndrome similar to hepatopulmonary syndrome has been described in congenital disorders without liver disease where either hepatic venous blood flow does not reach the lung or portal venous blood reaches the inferior vena cava without passing through the liver61,62. This supports the notion that factors either produced or metabolised in the liver influence the pulmonary microcirculation. Nevertheless, these factors have not been identified in humans so far. Clinical data suggest that enhanced release of nitric oxide in the lungs plays a pivotal role in the pathogenesis of the hepatopulmonary syndrome63. Increased pulmonary production of nitric oxide has been implicated in the development of vasodilation in patients with cirrhosis with hepatopulmonary syndrome. Increased concentrations of exhaled nitric oxide are positively correlated with the increase of alveolo-arterial oxygen difference64. However, the cause of the increased pulmonary nitric oxide production and its relationship with portal hypertension, the hyperdynamic circulation and the degree of liver injury remain undefined. In addition, the endothelin system has been implicated in the pathogenesis of hepatopulmonary syndrome.
Histological examination reveals dilated intrapulmonary arterioles and capillaries and dilated vascular channels between pulmonary arteries and veins. A typical finding in hepatopulmonary syndrome is orthodeoxia (arterial deoxygenation improving in recumbency), which leads to the debilitating clinical symptom of orthodeoxia-platypnea (hypoxemia and dyspnoea induced or worsened in the upright position). This phenomenon is explained by the worsening of diffusion-perfusion matching and an increase of the shunt fraction in the upright position because of increased perfusion of the lower lobes33. Recently Gomez et al65 published new set of data that corroborate this conviction: they recommend a PO2 decline >= 5% or >= 4 mmHg when upright position is assumed to diagnose the presence of orthodeoxia in hepatopulmonary syndrome.
Clinical features
The clinical relevance of the presence of hepatopulmonary syndrome has increased over the last decade likely due to a number of factors. Hepatopulmonary syndrome is characterized by a clinical triad that includes liver disease and/or portal hypertension, abnormal arterial oxygenation, and the presence of intrapulmonary vascular dilatations66. Patients with hepatopulmonary syndrome complain of progressive dyspnoea and can become increasingly cyanotic. Classically, an increase in dyspnoea with standing (platypnea) has been described, attributed to the predominance of vasodilation in the lung bases. However, the frequency or usefulness of this observation in the diagnosis of hepatopulmonary syndrome is undefined. Orthodeoxia is not unique to hepatopulmonary syndrome, but is highly suggestive of it. The hypoxemia may range from mild to profound, and its severity does not always correlate with the severity of the underlying liver disease. In addition, poor physical conditioning, smoking, ascites and/or intrinsic lung disease are often present and may also cause dyspnoea. Some patients develop clubbing of the fingers, and skin teleangectasias (spider angiomas) are typically seen in high number67. However, these features have not been prospectively investigated as indicators of the presence of hepatopulmonary syndrome. Physical examination might reveal evidence of liver disease, but findings in the lungs and the heart are generally normal unless coexisting disease is present.
A recent prospective clinical study by Schenk et al68, showed that the presence of hepatopulmonary syndrome has a major influence on survival in patients with cirrhosis. In this study, patients with hepatopulmonary syndrome showed a 3.8-times lower median survival time compared with those without this syndrome. In addition, a subgroup analysis according to the severity of liver disease evaluated with Child-Pugh score, showed a clear survival disadvantage in patients in Child class C.
Diagnosis
The diagnosis of hepatopulmonary syndrome is based on the concomitance of arterial deoxygenation and intrapulmonary vasodilatation in patients with liver disease and pulmonary symptoms. Arterial blood gases are used to detect gas exchange abnormalities in hepatopulmonary syndrome. Although arterial hypoxemia is arbitrarily defined as an arterial pO2 of less than 70 mmHg, diagnostic criteria for hepatopulmonary syndrome frequently include a widened alveolar arterial oxygenation gradient (> 20 mmHg) without hypoxemia as sufficient to indicate the presence of gas exchange abnormalities2. Nevertheless, the clinical importance and specificity of mild arterial blood gas changes are not well defined. In addition, the alveolar arterial oxygen gradient normally widens with age and should thus be corrected for age to avoid an overestimation of the prevalence of HS. Determination of alveolar arterial oxygenation gradient, by also taking into account the determination of pO2, allows a more accurate assessment of abnormal arterial oxygenation because the use of pO2 alone may underestimate the true degree of hypoxemia, which may be masked by the hyperventilation and hyperdynamic circulation that occurs in cirrhotic patients. Since orthodeoxia is a typical finding in hepatopulmonary syndrome, blood gas analyses should be obtained with patient in erect and supine positions. When abnormalities in gas exchange are detected, chest radiography and pulmonary function tests are generally performed to evaluate the presence of intrinsic cardiopulmonary disease and testing to detect intrapulmonary vasodilation is warranted. Pulmonary function testing may show a low diffusion capacity for carbon monoxide. However, this finding is not a prerequisite for the diagnosis of hepatopulmonary syndrome36. In all patients who have chronic liver disease and hypoxemia, the diagnosis of hepatopulmonary syndrome requires documentation of intrapulmonary vascular dilatation. Contrast echocardiography, lung perfusion scanning and pulmonary angiography are used to detect intrapulmonary vasodilation. Two-dimensional transthoracic contrast echocarhocardiography is used as a contrast agent. The contrast medium is visualised in the left atrium between three and six heart cycles after intravenous injection, when shunts typical of the hepatopulmonary syndrome are present69-71. However, an immediate visualization of injected contrast in the left heart indicates the presence of an intracardiac shunting, that is an important differential diagnostic element. Echocardiography performed by the transthoracic approach, when a good sonographic window is available, is more sensitive than lung perfusion scanning in detecting the presence of intrapulmonary vasodilation. Transesophageal contrast echocardiography by the endoscopic approach may amplify the sensitivity of detecting intrapulmonary vasodilation compared to transthoracic echocardiography because it ameliorates ultrasound imaging of the heart and may be helpful in identifying early hepatopulmonary syndrome that is missed by transthoracic echocardiography72. Contrast echocardiography gives positive findings in up to 40% of cirrhotics with normal arterial blood gases, suggesting that a mild degree of intrapulmonary vasodilation, insufficient to alter gas exchange and cause hepatopulmonary syndrome, is a common finding in cirrhosis. Radionuclide lung perfusion scanning, using technetium-labeled macroaggregated albumin particles (99mTc), is also commonly employed to detect intrapulmonary shunting. In the presence of a cardiac right-to-left shunt or intrapulmonary vascular dilatation the uptake of 99mTc macroaggregated albumin can be documented in other organs such as the brain or the spleen. An advantage of this technique, is that a positive lung scan is specific for the presence of hepatopulmonary syndrome even in the setting of co-existent intrinsic lung disease. This technique has been employed in the diagnostic workout of hepatopulmonary syndrome and for the quantification of the extent of shunting73. Additionally, it is also useful for following the progression and/or resolution of disease. Nevertheless, radionuclide lung perfusion scanning is less sensitive than contrast echocardiography in detecting intrapulmonary vasodilation. Therefore, the radionuclide lung perfusion scan is not an optimal screening test for hepatopulmonary syndrome, but is a good complementary tool. Pulmonary angiography is not a standard diagnostic tool in patients with evidence of hepatopulmonary syndrome. It is an invasive and insensitive diagnostic technique for revealing intrapulmonary vasodilation, and again it is not useful as a screening test. However, in patients in whom the response to oxygen administration is scarce, condition which suggests right-to-left shunting, pulmonary angiography can be performed to identify focal arteriovenous malformations that may be amenable to transcatheter embolisation74. In figure 1 the diagnostic flow-chart of hepatopulmonary syndrome is shown.
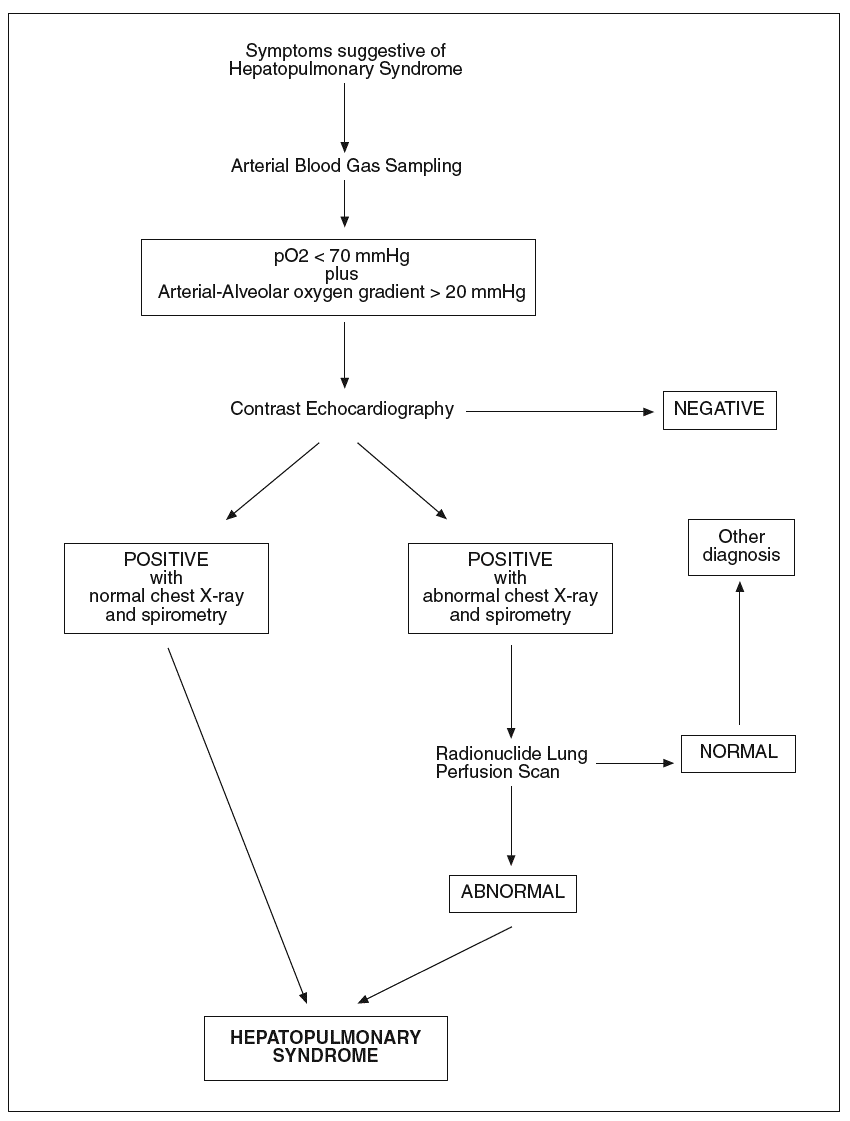
Fig. 1. Diagnostic flow-chat forhepatopulmonary syndrome.
Therapy
The treatment of hepatopulmonary syndrome includes the correction of hypoxemia by administration of oxygen. However, in severe cases and when right-to-left shunting is present, hypoxemia might not be fully correctable. Theoretically, the ideal treatment of hepatopulmonary syndrome would consist of a drug or any other means to reverse the intrapulmonary vascular dilatation. No clearly effective medical therapies for hepatopulmonary syndrome have been found. Somatostatin, almitrine, indomethacin, L-NAME and plasma exchange have all been tried unsuccessfully in a study performed by Abrams et al45. There is a report by Song et al75, that aspirin increased arterial oxygenation in 2 children with hepatopulmonary syndrome, and a case report and subsequent open label trial using garlic also suggested a beneficial effect76,77. Increased production of nitric oxide is a potential therapeutic target, but this approach has not been established as a conventional approach. Diets containing low amounts of L-arginine, the substrate of nitric oxide synthase have provided no long-standing benefits. One case report and a recent study by Schenk et al78, have found that the intravenous infusion of methylene blue, an inhibitor of guanylate cyclase, which mediates the intracellular effects of nitric oxide, causes pulmonary vasoconstriction and reduction of hypoxemia in patients with hepatopulmonary syndrome. These reports highlight the need to target likely pathogenetic mechanism in randomised multicenter trials. A single case report by Anel et al79, suggests that norfloxacin (by acting on macrophages as previously mentioned) may have contributed to improvement in oxygen saturation in hepatopulmonary syndrome.
In recent years the role of TIPS has been anecdotically evaluated. Six available case reports have reported the results obtained by means of TIPS insertion on hepatopulmonary syndrome in cirrhosis, and have suggested that TIPS may be of value to improve gas exchange in this syndrome. However, too short duration of follow-up in 2 patients, the presence of concomitant hepatic hydrothorax and an unusual presentation of hepatopulmonary syndrome with rapid onset of profound hypoxemia in decompensated liver disease with minimal portal hypertension, do not allow to draw any conclusion on the potential utility of TIPS in this clinical condition. Nevertheless, another report by Allgaier et al80, clearly demonstrated an increase in arterial oxygenation of 20 mmHg six months after TIPS placement despite persistence of significant intrapulmonary shunting, based on radionuclide lung perfusion scanning. These findings suggest that improved oxygenation was not due to reversal of intrapulmonary vasodilation but more likely due to a redistribution of pulmonary blood flow to normal ventilation to perfusion ratio regions induced by an increase in cardiac output induced by the procedure. There is a report of failure of TIPS to improve oxygenation in one patient and identification of two patients81 where hepatopulmonary syndrome developed in the setting of a functioning TIPS. These findings suggest that TIPS, at least at present, should be considered an experimental treatment for hepatopulmonary syndrome.
In patients with progressive and refractory hypoxemia, liver transplantation is the treatment of choice since it can correct most of the circulatory, neurohumoral and pulmonary underlying abnormalities secondary to the correction of the underlying chronic liver disease. Liver transplantation is the only proven therapy for hepatopulmonary syndrome based upon the total resolution or significant improvement in gas exchange post-operatively in more than 85% of reported series82. However, improvement of hypoxemia after liver transplantation is not an early phenomenon and several months or even years may be required before resolution. In addition, mortality is increased after transplantation in patients with hepatopulmonary syndrome compared to subjects without hepatopulmonary syndrome. Innovative approaches such as frequent body positioning or inhaled nitric oxide have been used to improve post-operative gas exchange.
On the basis of available current experimental and human data, there are several potential therapeutic targets for medical therapies in hepatopulmonary syndrome. Inhibiting the sequence of events involved in TNF-a overproduction and macrophage activation through selective intestinal decontamination (pentoxiphylline, anti-TNF antibodies) would appear to be an attractive therapeutic target. Modulating pulmonary endothelin action or nitric oxide production could also be beneficial. Blocking the production and/or release of endothelin and other potential mediators, from their splanchnic source might prevent or ameliorate the cascade of events leading to intrapulmonary vasodilation.

