



INTRODUCCION
Reiteradamente vemos que este concepto, síndrome autoinflamatorio o enfermedad autoinflamatoria sistémica, aparece entre los temas de actualidad que se consideran importantes en las reuniones científicas. Lo vemos también en las revistas nacionales que recibimos los reumatólogos e incluso algunos de nuestros pacientes nos preguntan por ellos, o mejor dicho, por alguno de ellos, porque le han sugerido que su hijo/a quizá lo presente. También es cierto que cuando, con gran interés por nuestra parte y la mejor buena voluntad, hemos asistido a alguna de las sesiones sobre este tema, hemos salido con la impresión de que apenas habíamos entendido un diez por ciento del contenido y que lo que nos han contado sobre la fiebre mediterránea familiar ya lo sabíamos. Y es verdad, porque la clínica de la fiebre mediterránea familiar (FMF) no se ha modificado en los últimos siglos. Ahora, ¿por qué este interés en este grupo de enfermedades?, ¿por qué hay laboratorios clínico-básicos que dedican casi el total de su actividad a estudiar a estos pacientes? Responder a estas preguntas es el objetivo de esta revisión que no pretende ser exhaustiva ni tampoco dejar exhausto al que la lea, sino más bien clarificar conceptos, intentar hacer lo complicado simple, sin, por supuesto, desmerecer las excelentes revisiones ya publicadas sobre el tema1-3.
¿QUÉ SON?
En los últimos 5 años se ha individualizado cerca de una decena de síndromes que guardan cierta similitud con la FMF o con la artritis idiopática juvenil de comienzo sistémico (sAIJ) en su presentación clínica, pero en los que el trastorno inflamatorio se diferencia de estas entidades tanto en su origen como en su patrón evolutivo, y en los que se mantiene un estado inflamatorio continuado sin que exista, o al menos hayamos sido capaces de detectarla, la producción de ningún autoanticuerpo determinado ni tampoco linfocitos antígeno-específicos. De aquí que no hablemos de enfermedad autoinmunitaria sino de síndrome autoinflamatorio o, como hemos mencionado al principio, de enfermedad autoinflamatoria sistémica. En todos ellos el aparato locomotor es uno de los órganos diana de la enfermedad.
Entre las diferentes entidades clínicas englobadas en los síndromes autoinflamatorios, la presente revisión tratará de aquellas cuyas bases genéticas han sido identificadas, y de las que, a pesar de ser consideradas como enfermedades raras por su incidencia, en nuestra población se ha diagnosticado a pacientes afectados de cada una de ellas, sin presentar relación con ningún origen étnico especial.
¿POR QUÉ SE PRODUCEN?
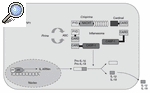
Vamos a fijarnos por un momento en el esquema representado en la figura 1. Aquí se esquematiza lo que se ha denominado inflamasoma. Al contrario de lo que ocurre con organelas citoplasmáticas que pueden tener nombres similares (p. ej., lisosoma), el inflamasoma no ocupa un lugar determinado y fijo ni está delimitado por ninguna membrana. Está constituido por una serie de proteínas cuya función fundamental es regular el paso de pro-IL-1ß a IL-1ß (aunque interviene también en el procesamiento de pro-IL-18 y pro-IL-33), y de esta manera iniciar la cascada inflamatoria. Dentro de este conjunto de proteínas, la que directamente realiza la conversión de pro-IL-1ß a IL-1ß es la caspasa-14,5. ¿Esta información es importante para el tema que intentamos desarrollar? Sí, porque en todas las enfermedades autoinflamatorias se da una regulación al alza (up-regulation) del proceso de conversión de la forma activa de IL-1ß.
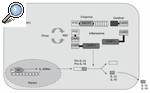
Figura 1
Esquema del inflamasoma. Producción de la forma madura de IL-1ß e IL-18.
ASC: apoptosis associated speckle-like protein containing CARD; CARD: caspase recruitment domain; CASP-1: caspasa 1; PYD: pyrin domain.
Todas las proteínas en cursiva están relacionadas con la aparición de un síndrome autoinflamatorio (véase el texto).
Tomemos como ejemplo la FMF. En el año 1997 los resultados de 2 grupos de investigadores diferentes establecieron que mutaciones en un gen anteriormente no descrito, al que denominaron MEFV (por MEditerranean FeVer), estaban asociadas con la FMF6,7. Dicho gen está localizado en el brazo corto del cromosoma 16, y codifica para una proteína de 781 aminoácidos de longitud, que recibió 2 nombres diferentes: pirina o marenostrina. En la actualidad se ha identificado más de 80 mutaciones asociadas con la enfermedad, la gran mayoría de las cuales se localizan en el exón 10 del gen, y que están disponibles en la base de datos internacional INFEVERS (http://fmf.igh.cnrs.fr/infevers). No se conoce con profundidad la función de la proteína pirina/marenostrina (P/M), aunque se postula que actúa como regulador negativo del inflamasoma. Dicha regulación negativa se llevaría a cabo mediante el secuestro de la proteína ASC (apoptosis-associated speck-like protein containing a CARD) por la proteína P/M, mediante interacciones entre los dominios pirina (PyD) existentes en ambas proteínas, lo que daría lugar a una disminución de la actividad caspasa-1 y, en consecuencia, a una disminución de las formas activas de IL-1ß. Las diferentes mutaciones descritas hacen que la proteína P/M pierda esta función de regulación negativa y consecuentemente se mantenga o aumente la producción de IL-1ß.
Pongamos un segundo ejemplo: la acción de la criopirina (proteína determinante de los síndromes periódicos asociados a criopirina (CAPS) (fig. 1). En los síndromes CAPS se cree que las mutaciones del gen producen una criopirina funcionalmente "hiperactiva", que se traduce en una activación aumentada de la caspasa-1, de tal manera que se ha podido demostrar en estos pacientes un aumento importante de la IL-1ß generada. Esta citocina sería la causa directa de la fiebre, al actuar sobre el centro termorregulador, y de la leucocitosis y la neutrofilia al actuar sobre los precursores hematopoyéticos de la médula ósea. Asimismo, la IL-1ß genera un aumento de la IL-6 y, en consecuencia, es causante de forma indirecta de las lesiones cutáneas y de la trombocitosis. Así, en los síndromes CAPS el resultado es el mismo, un aumento en la producción de IL-1ß, pero la causa es diferente: aquí hay una regulación al alza y no una pérdida de función de control negativo8.
Aunque queda fuera del objetivo de esta revisión, de la misma forma que hemos explicado cómo los errores genéticos en la FMF o en los síndromes CAPS modifican la producción de citocinas proinflamatorias, podríamos poner en relación todos y cada uno de los errores genéticos de los diferentes síndromes autoinflamatorios con alteraciones en la función del inflamasoma.
¿CUALES SE DIAGNOSTICAN EN LA EDAD PEDIATRICA Y CUALES EN EL ADULTO?
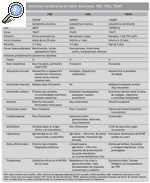
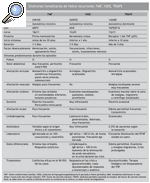
Antes de contestar a esta pregunta haremos un breve resumen de la presentación clínica de las diferentes entidades. Tenemos que tener en cuenta que con el término de síndromes hereditarios de fiebre recurrente se engloban la FMF, el síndrome de hipergammaglobulinemia D asociado a fiebre periódica (HIDS) y el síndrome periódico asociado al receptor de TNF* (TRAPS). El término CAPS engloba el síndrome CINCA/NOMID, el síndrome de Muckle-Wells y el síndrome autoinflamatorio familiar inducido por el frío (FCAS); las granulomatosis sistémicas infantiles agrupan el síndrome de Blau y la sarcoidosis de inicio temprano; el síndrome de artritis piogénica esteril, pioderma gangrenoso y acné (PAPA) tiene una presentación clínica y anomalía genética diferentes.
La FMF9,10 es el más común y el mejor conocido de los síndromes que cursan con fiebre recurrente, de herencia autosómica recesiva y que afecta, sobre todo, a las poblaciones descendientes de judíos sefarditas y askenazí, armenios, árabes y turcos. Tiene 2 formas sindrómicas de presentación: FMF de tipo I, la más frecuente, y la que resumiremos a continuación, y FMF de tipo II, en la que se engloba a los pacientes que nunca han presentado ataques de fiebre ni de ninguno de los otros síntomas asociados, a quienes se diagnostica en etapas relativamente tardías de la vida por amiloidosis secundaria (habitualmente renal). La mayor parte de los pacientes presentan la FMF de tipo I, con inicio de los primeros síntomas durante la edad escolar o adolescencia; en más del 80% de los casos el primer brote tiene lugar antes de los 20 años (1).
La forma habitual de presentación se inicia como brotes de fiebre, acompañados de dolor abdominal, dolor torácico secundario a pleuritis y/o artralgias. Mientras éstas son las localizaciones inflamatorias más comunes, los pacientes pueden referir síntomas en relación con cualquier otro órgano de la economía, especialmente la piel y los órganos linfoides (ganglios regionales y bazo). Los brotes pueden ser desencadenados por la menstruación en las niñas adolescentes tras la menarquia, pero también por el estrés emocional o el agotamiento físico.
Algunos pacientes pueden experimentar síntomas prodrómicos antes de iniciarse el brote, como escalofríos intensos que persisten durante horas. Sin embargo, la fiebre, de hasta 39-40 °C, comienza de forma brusca, se mantine entre 12 y 72 h, y se acompaña de los síntomas referidos. Estos síntomas secundarios varían de un paciente a otro, e incluso en el mismo paciente, de una ataque a otro. Todos los patrones de fiebre se han descrito en la FMF: oscilante, persistente, mantenida.
Entre un 50 y un 70% de los pacientes con FMF refieren dolor abdominal como primer síntoma de su enfermedad. La artralgia/artritis se presenta en el 75% de los judíos del Norte de África (menos del 50% en las otras poblaciones). Es característicamente monooligoarticular y afecta a grandes articulaciones: rodillas, tobillos, caderas. Algunos pacientes pueden experimentar dolor sacroilíaco, no siendo portadores de HLA-B27. La piel puede verse afectada por lesiones purpúricas, o bien por placas erisipeloides, localizadas con mayor frecuencia en la región pretibial, eritematosas, calientes y dolorosas al tacto.
Es un dato común en todos los estudios que el dolor abdominal, la artralgia/artritis y estas lesiones cutáneas que recuerdan a la erisipela sean más frecuentes en la infancia que en la edad adulta.
Hipergammaglobulinemia D asociada a fiebre periódica11-14
La primera descripción detallada de este síndrome fue publicada por Van der Meer et al11 en 1984, y fue seguida por diversas series que han comunicado los hallazgos clínicos y genéticos en estos pacientes.
La clínica comienza habitualmente durante el primer año de vida (en muchas ocasiones antes de los 6 meses). Los accesos febriles se mantendrán durante toda la vida del paciente, si bien es en la infancia cuando su frecuencia es mayor, aunque en la edad adulta puede estar asintomático durante años. Los ataques son desencadenados por infecciones, vacunas, traumatismos menores, estrés o cirugía. La fiebre se instaura de forma brusca, en forma de picos de hasta 40 °C, y se acompaña característicamente de: adenopatías laterocervicales bilaterales (86-94%) y dolorosas a la palpación, dolor abdominal difuso (72-86%), vómitos (73-82%), diarrea (56-86%), cefalea (52%), poliartralgias (80%) y lesiones cutáneas (73-82%)9,10. La afectación de la piel es común y las lesiones, en ésta, polimorfas: maculares y difusas, que afectan a palmas y plantas; papulares; urticariales; nodulares; purpúricas o acneiformes. Las úlceras orales y/o genitales no son infrecuentes en el síndrome HIDS. La biopsia cutánea muestra habitualmente cambios discretos compatibles con vasculitis.
Síndrome periódico asociado al receptor de TNF* (TRAPS)15
La edad media de comienzo son los 3 años (edad preescolar). Los ataques son más prolongados que en las 2 enfermedades anteriores, con una duración de hasta 3 semanas, y se repiten con una cadencia variable (4-6 semanas). Los pacientes describen los síntomas prodrómicos como un comienzo progresivo de dolor muscular profundo, que va en aumento en el curso de 2-3 días hasta alcanzar su máxima intensidad y disminuye posteriormente también de forma progresiva. La fiebre está siempre presente en los casos pediátricos, pero puede faltar en los adultos. El síntoma diferenciador más claro del TRAPS es la mialgia, que afecta característicamente sólo a un área corporal, y aumenta y disminuye en intensidad a lo largo de los días en los que se establece el ataque. Cuando la mialgia afecta a un área correspondiente a una articulación, puede haber derrame articular y contractura transitoria periarticular y de la extremidad afectada.
Los hallazgos cutáneos también son diferentes en esta entidad: aparece una placa eritematosa, focal, situada sobre el área de dolor muscular que, durante el episodio inflamatorio, se desplaza de forma centrífuga, y puede llegar a tener varios centímetros de diámetro. Estas placas eritematosas migratorias son dolorosas a la palpación, están calientes y palidecen a la presión. Sin embargo, la lesión cutánea del TRAPS también puede adquirir formas diversas a la referida: eritema generalizado, lesiones urticariales, placas serpinginosas y otras.
Como en los otros síndromes, el dolor abdominal es frecuente (hasta en un 92%), así como vómitos, estreñimiento, fenómenos de oclusión o seudooclusión intestinal. El 82% de los pacientes presentan conjuntivitis, edema o dolor periorbitario, en algunas ocasiones como pródromos de los episodios inflamatorios. El dolor torácico, de causa reumática o pleurítica está presente en un 57% de los casos. El dolor testicular o escrotal puede darse pero no es especialmente frecuente, así como tampoco la aparición de linfadenopatías.
Estos datos clínicos referidos hasta el momento sobre los síndromes hereditarios de fiebre recurrente se ha intentado esquematizarlos en la tabla 1. Podemos decir ahora que esperamos ver a pacientes con HIDS en niños con edad inferior al año de vida (lactantes), mientras que los síntomas relacionados con FMF o TRAPS se inician en la edad escolar, adolescencia o comienzo de la edad adulta (2).
Síndrome CINCA/NOMID16,17
En 1981, los reumatólogos pediatras que trabajaban en el Hôpital Enfants Malades en París (Francia) describieron los hallazgos clínicos y analíticos de 3 pacientes, no consanguíneos, que asociaban a artritis y crisis febriles un exantema máculo-papular generalizado, meningitis crónica, lesiones oculares y retraso mental, y que ellos pensaron que, si bien se asemejaban a los pacientes con AIJ sistémica, no presentaban la misma enfermedad; les debemos a ellos haber podido desligar del común de pacientes con artritis juvenil rebelde al tratamiento, sólo por las características clínicas particulares, un cuadro autoinflamatorio que posee alteraciones genéticas propias que se descubrirían 20 años más tarde. Hasta hoy, se han identificado unos 100 casos en el mundo entero.
CINCA proviene del acrónimo chronic infantile neurologic cutaneous and articular syndrome, así como NOMID lo hace de neonatal-onset multystem inflammatory disease.
1. Los signos cutáneos suelen aparecer ya en los primeros días de vida: la erupción es bastante similar en todos los pacientes, se trata de lesiones urticariformes recurrentes, poco o nada pruriginosas, cambiantes de un día a otro e incluso a lo largo de un mismo día.
2. La afectación articular, sin embargo, es muy diferente de un paciente a otro. Hay formas poco agresivas que se mantienen durante años, como artralgias y, sólo ocasionalmente, se acompañan de inflamación articular sin dejar ninguna secuela al terminar el crecimiento. En el otro extremo, están las formas en las que la artropatía se inicia de forma muy precoz, de forma simétrica, que afecta a metáfisis y epífisis. Es característica la aparición de un sobrecrecimiento rotuliano: osificación precoz de la rótula, aspecto irregular (en miga de pan) de la trama ósea epifisaria tibial y rotuliana, desorganización del cartílago de crecimiento de la extremidad superior de la tibia. Estas lesiones son casi patognomónicas, y permiten confirmar el diagnóstico si otros elementos del síndrome están presentes.
3. La afectación del sistema nervioso central es un elemento importante del síndrome. Puede haberse iniciado en los primeros años de vida, pero no sospecharse como parte integrante de un síndrome: cefaleas, vómitos, crisis convulsivas, trastornos motores transitorios. La meningitis crónica es el hallazgo más común, en la que existe un aumento variable de celularidad en el líquido cefalorraquídeo, con un predominio de polimorfonucleares. Se tiende a poner en conexión la meningitis crónica con los principales hallazgos oculares.
4. Aspecto morfológico. Como ocurre en otros síndromes determinados genéticamente, todos estos pacientes poseen rasgos dismórficos comunes, lo que hace que guarden un parecido entre sí (sibling-like resemblance). Muchos de ellos son rubios, poseen una frente abombada, ojos sobresalientes, filtrum largo y labio superior fino. Las manos y los pies tienen un aspecto tosco, con dedos "en maza". En algunos de ellos, las palmas y plantas adquieren un aspecto como arrugado. En los adultos, la conformación en silla de montar de la raíz nasal se hace más evidente.
Síndrome de Muckle-Wells3,18
Muckle y Wells describieron en 1962 un cuadro clínico heredado de forma autosómica dominante, caracterizado por la presencia de lesiones urticariales crónicas, sordera neurosensorial progresiva y amiloidosis. Si bien los síntomas cutáneos y la fiebre se inician en la infancia, la sordera neurosensorial es una manifestación tardía, por lo que el diagnóstico se retrasa con frecuencia a los primeros años de la edad adulta. La gravedad de la enfermedad reside en la frecuencia con la que ésta asocia amiloidosis: un 25% de los casos. Este hecho contrasta con lo que ocurre en otras enfermedades inflamatorias crónicas, en las que el porcentaje de pacientes con esta complicación se mantiene bastante constante alrededor de un 5%.
Síndrome autoinflamatorio familiar inducido por el frío (FCAS)3
Tras la exposición al frío ambiental, el paciente desarrolla exantema urticarial generalizado, seguido de dolor e inflamación de las articulaciones, escalofríos y fiebre. Estos síntomas se hacen más evidentes al comienzo de la edad adulta. El espectro clínico, sin embargo, es muy amplio, con pacientes en los que nunca aparecen las lesiones cutáneas y/o la fiebre inducidas por el frío, sólo el dolor articular, mientras que en otros los episodios febriles son severos, acompañados de artritis, y se ha descrito cardiomiopatía inflamatoria, nefropatía y tiroiditis normofuncionante. Esta entidad se diferencia de la urticaria física inducida por el frío por la demora entre la exposición generalizada al frío y la aparición de la clínica, así como por un test del cubito de hielo negativo.
Por tanto, de los síndromes autoinflamatorios relacionados con alteraciones en el gen CIAS1 y la proteína criopirina, el síndrome CINCA/NOMID es el síndrome característico del niño, mientras que el diagnóstico de los síndromes de Muckle-Wells y FCAS suele retrasarse hasta los primeros años de la edad adulta.
Las granulomatosis sistémicas infantiles, por definición, son enfermedades pediátricas, con un comienzo antes de los 4 años de edad. Se caracterizan por la presencia de la tríada artritis+uveítis+exantema. La artritis es la manifestación más frecuente. Se caracteriza por ser una poliartritis crónica simétrica, y afecta tanto a grandes como pequeñas articulaciones, moderadamente dolorosa, con un importante componente de sinovitis que da lugar a grandes formaciones quísticas (cystic swelling) sobre las articulaciones afectadas. Sólo se acompaña de una ligera molestia a la presión y la limitación del movimiento se produce como una complicación tardía. La uveítis puede afectar tanto a cámara anterior como a cámara posterior y ser extremadamente grave y rebelde al tratamiento. El glaucoma es una complicación ocular común. De las 2 entidades, el síndrome de Blau corresponde a la forma familiar19,20, mientras que la sarcoidosis de inicio temprano se produce como casos esporádicos, sin familiares afectados, como consecuencia de mutaciones de novo.
El síndrome PAPA, caracterizado por artritis piogénica estéril, pioderma gangrenoso y acné quístico, constituye una entidad rara identificada en unas 25 familias hasta el momento. Durante la edad escolar, los pacientes presentan fundamentalmente la clínica articular, y durante la adolescencia se ponen de manifiesto el acné quístico y el pioderma gangrenoso. Es por ello que el diagnóstico de la enfermedad se realiza habitualmente en la adolescencia y/o en la edad adulta.
En resumen:
Entidades de diagnóstico en el período neonatal/ lactante: síndrome CINCA/NOMID, HIDS.
Entidades de diagnóstico en la edad preescolar/escolar/adolescencia: FMF, TRAPS, síndrome de Muckle-Wells, FCAS, síndrome PAPA.
Entidades de diagnóstico en la edad adulta: FMF, síndrome de Muckle-Wells.
¿COMO CONFIRMAR EL DIAGNOSTICO?
Nos podría parecer que, dado que conocemos cuál es el gen afectado en cada uno de los síndromes autoinflamatorios (tabla 2), y qué mutaciones son causa de la enfermedad, el diagnóstico se debería basar en la confirmación, mediante estudio genético, del hallazgo de alguna de estas mutaciones. Sin embargo, la experiencia acumulada durante estos últimos años ha puesto de manifiesto que no podemos aplicar este criterio debido a la heterogeneidad clínica y genética en estas enfermedades, lo cual se traduce en que hay pacientes con clínica característica sin mutaciones y, al contrario, familiares de los pacientes que, siendo portadores de una mutación causante de enfermedad, no tienen síntomas clínicos.

Tomemos como ejemplo, de nuevo, la FMF. En la actualidad han sido identificadas más de 80 mutaciones asociadas con la enfermedad, la gran mayoría de las cuales se localizan en el exón 10 del gen MEFV. Entre estas mutaciones, 4 de ellas (M680I, M694V, M694I, V726A) reciben el nombre de mutaciones fundadoras, por ser las mutaciones más frecuentemente detectadas en las poblaciones denominadas ancestrales, con una alta prevalencia de la enfermedad (judíos, turcos, árabes y armenios). Se da la circunstancia de que dichas mutaciones fundadoras son también las mutaciones más frecuentemente detectadas en pacientes afectados de FMF pertenecientes a poblaciones diferentes de las ancestrales, posiblemente como consecuencia de los flujos migratorios llevados a cabo por la humanidad en los dos últimos milenios, que han generado la dispersión de dichas variantes genéticas. Sin embargo, desde un punto de vista clínico, a pesar de que clásicamente la FMF se cataloga como una enfermedad autosómica recesiva, existe un porcentaje importante de pacientes clínicamente diagnosticados de FMF que bien no portan ninguna mutación en el gen MEFV o bien portan sólo una (heterocigosis), y en estos últimos, por tanto, la enfermedad se comporta como si fuera dominante. Para intentar explicar por qué ocurre esto, diferentes grupos han propuesto que el gen MEFV sería sólo uno de los diversos genes asociados a la FMF, si bien los otros genes, que actuarían como moduladores-modificadores de MEFV, están, al día de hoy, pendientes de ser identificados21.
Lo mismo ocurre para el síndrome CINCA/NOMID. Mientras tras las primeras publicaciones22 se pensaba que todos los pacientes portaban mutaciones en el gen CIAS1, hoy sabemos que sólo se encuentran mutaciones en el 50% de los niños que clínicamente presentan la enfermedad, y se desconoce la base molecular subyacente en el resto de los casos.
En resumen, si el análisis genético confirma nuestra sospecha clínica, el diagnóstico es seguro. Sin embargo, el razonamiento no puede aplicarse a la inversa: la falta de confirmación genética no invalida el diagnóstico clínico. Esta afirmación, que está claramente demostrada para FMF y CINCA, debe tenerse en cuenta también al considerar el diagnóstico del resto de los síndromes autoinflamatorios.
¿ALGUN TRATAMIENTO ES EFICAZ?
Al hablar de tratamiento, hay que tener en cuenta que la FMF no se comporta como el resto de los síndromes autoinflamatorios, ya que responde en la mayoría de los casos a un fármaco clásico y bien conocido: la colchicina. Haremos un pequeño resumen sobre el esquema a seguir en la FMF antes de pasar al resto de las entidades mencionadas.
En la FMF el tratamiento con colchicina oral, administrado diariamente, fue evaluado en estudios a doble-ciego en la década de los años setenta. El tratamiento estándar se estableció en 1 mg/día, aunque los autores defienden una dosis óptima para el adulto entre 1,5 y 2 mg/día. De la misma manera, recomiendan ajustar la dosis para la superficie o el peso corporal (0,45 mg/m2/día o 0,03 ± 0,02 mg/kg/día) en el paciente pediátrico. Los niños menores de 5 años son especialmente "metabolizadores rápidos" de cualquier fármaco y, en el caso de la colchicina, pueden necesitar hasta 0,07 mg/kg/día. Habitualmente repartimos la dosis total diaria en 2 o 3 tomas. Sólo con colchicina se consigue una remisión de los síntomas en el 70% de los casos, mientras que un 20-25% experimentan una reducción en el número de ataques y de su gravedad, pero no la remisión completa23 y en un 5% de ellos los síntomas no se modifican en absoluto. Sin embargo, aun en éstos, la colchicina es capaz de prevenir la aparición de amiloidosis y de sus consecuencias renales. Puede ser utilizada en el lactante, a partir del año de vida. Sus efectos secundarios son menores comparados con el gran beneficio que comporta en los pacientes con FMF; el más frecuente es la diarrea secundaria a la intolerancia a la lactosa (creada a su vez por el fármaco).
En el 30% de los pacientes que sólo alcanzan un control parcial de los síntomas, se han utilizado otros tratamientos como: interferon-alfa, talidomida, azatioprina, y más recientemente agentes anti-TNF*, especialmente infliximab24. Los resultados prometedores de estos últimos no nos permiten concluir aún sobre su efectividad en el control a largo plazo de la amiloidosis secundaria. Algunos autores han utilizado anti-IL-1ra con buenos resultados (comunicación personal).
En el síndrome HIDS, sólo algunos pacientes responden positivamente al tratamiento con corticoides, especialmente en los brotes pero no tanto en su prevención. Un ensayo clínico con simvastatina (inhibidor de la enzima HMG-CoA reductasa) llevó a una disminución en el número e intensidad de las crisa como una buena alternativa terapéutica25, y se ha utilizado hasta el momento en la dosis habitual para la AIJ (queda por tanto saber si la forma de administración y la dosis deberían ajustarse mejor para el tratamiento del síndrome HIDS). De nuevo, hay casos aislados de tratamiento anti-IL-1ra con buena respuesta26.
De igual modo, en el síndrome TRAPS los glucocorticoides son capaces de disminuir la intensidad de los síntomas, pero no de reducir la frecuencia de los brotes. Se recomienda una dosis de 1 mg/kg/día de prednisona en el día en que comienza el brote, y disminuirla progresivamente en los 7-10 días siguientes. Los resultados de un estudio piloto con 7 pacientes, con etanercept, son prometedores en cuanto al control de los síntomas en las crisis y el efecto ahorrador de corticoides27. Asimismo, al igual que ocurre con los corticoides, etanercept no es capaz de abolir por completo la aparición de las crisis febriles ni de los procesos inflamatorios subclínicos)28.
Los antiinflamatorios no esteroideos pueden aliviar el dolor, aunque no cambien el curso de la enfermedad en el síndrome CINCA/NOMID. Los glucocorticoides, reducen la fiebre y el dolor, pero no son eficaces en cuanto a las lesiones cutáneas, del sistema nervioso central o articulares. La utilización de inmunomoduladores eficaces en la AIJ no ha resultado en un mayor beneficio en el caso del síndrome CINCA/NOMID. Recientemente, se han comunicado respuestas clínicas, hematológicas y bioquímicas positivas en pacientes tratados con bloqueadores de la IL-1, mediante el empleo de anakinra. Estas respuestas positivas se han objetivado tanto en pacientes con síndrome CINCA/NOMID portadores de mutaciones en el gen CIAS1 como en pacientes sin mutaciones en este gen29,30. Según los datos de investigación actuales es posible que, en un futuro, otros agentes terapéuticos que bloquean la acción de IL-1 se pueda utilizarlos también en el tratamiento del síndrome CINCA/NOMID. Lo referido para este síndrome es trasladable a los otros síndromes que dependen de la criopirina (Muckle-Wells, FCAS), si bien en el caso del síndrome autoinflamatorio inducido por el frío parece desproporcionado plantear tratamiento con agentes biológicos cuando la clínica se reduce drásticamente al evitar la exposición a bajas temperaturas.
Para el caso del síndrome de Blau/EOS no hay estudios controlados. La experiencia escasa de los distintos autores lleva a afirmar que los antiinflamatorios no esteroideos son poco eficaces. En el caso de la artritis, puede haber respuesta a los corticoides y se ha visto una respuesta excelente al tratamiento con infliximab. Sin embargo, el tratamiento anti-TNF* puede hacer empeorar el componente ocular. Hay cierta respuesta de la uveítis a la ciclosporina A.
Finalmente, en el PAPA sólo los pacientes más jóvenes pertenecientes a las familias portadoras del síndrome han sido tratados con fármacos inductores de remisión. De ellos, leflunomida parece ser especialmente eficaz. Hay publicaciones sobre casos aislados tratados con éxito con infliximab, etanercept y anakinra31-33.
Sintetizando la respuesta a la pregunta sobre el tratamiento diremos que:
El tratamiento clásico con colchicina sigue siendo de elección en la FMF.
En casi todos los síndromes, los corticoides son capaces de reducir los síntomas pero no de controlar la enfermedad.
Los nuevos agentes biológicos anti-TNF* y anti-IL-1 se muestran eficaces en el control clínico y analítico en la mayor parte de los pacientes tratados, especialmente en los síndromes dependientes de CIAS1/criopirina.
Esperamos haber cumplido con el objetivo de esta breve revisión: hacer una puesta al día, en forma esquemática, de lo que el reumatólogo general hoy debe conocer sobre los síndromes autoinflamatorios. El lector debería conocer más (y no menos), sobre ellos, al terminar de leer el artículo. De todas formas le estamos agradecidos por haber querido dedicar un poco de su tiempo a un grupo de enfermedades que afectan, en su inicio, y en la mayoría de los casos, a pacientes en edad pediátrica.
(1) Utilizaremos de ahora en adelante la subdivisión comúnmente usada en pediatría para referirnos a las distintas etapas del niño, desde su nacimiento hasta la edad adulta. Así: lactante de 0 a 2 años; edad preescolar de 3-5 años; edad escolar de 6-10/12 años; adolescencia de 10/12 a 16/17 años; adulto jóven de 17-21 años. Tomado de Nelson, Tratado de Pediatría, 1992. Aceptada por la Asociación Española de Pediatría.
(2) Nota del autor: querríamos hacer hincapié en que a lo largo del artículo y en las tablas nos estamos refiriendo siempre al inicio clínico de la enfermedad; es obvio, que en muchos casos habrá un lapso, más o menos prologando pero a veces de años, entre este inicio y el diagnóstico.