




Las miopatías congénitas son un grupo de trastornos musculares esqueléticos hereditarios, clínica y genéticamente heterogéneos, definidos de acuerdo con los hallazgos histopatológicas observados en las biopsias musculares. Las lesiones histopatológicas más comunes son los agregados de proteínas, los cores y el aumento de las centralizaciones nucleares. En el campo de las miopatías congénitas musculares se han logrado avances en los últimos años, al definir nuevos trastornos. En particular, el desarrollo de las técnicas de secuenciación de nueva generación o secuenciación masiva (NGS) permitieron la identificación de alrededor de 20 nuevas formas de miopatías congénitas. Se han logrado algunos avances en el conocimiento de la alteración de la contractilidad muscular en el campo de las miopatías nemalínicas. Con la perspectiva de futuros ensayos clínicos, el alcanzar un diagnóstico genético e histopatológico y establecer las medidas de atención aceptadas internacionalmente, son fundamentales para reducir la carga de la enfermedad en los pacientes con miopatías congénitas.
Congenital myopathies are a group of primary hereditary, clinically and genetically heterogeneous skeletal muscle disorders, defined according to histopathologic lesions observed in muscle biopsies. The most common histopathologic lesions are protein aggregates, cores, and increased nuclear centralizations. The field of muscle congenital myopathies has met progress in the recent years by defining new disorders. In particular, next generation sequencing (NGS) techniques allowed the identification of around 20 novel forms of congenital myopathies. Some insights on the alteration of muscle contractility have been gained in the field of nemaline myopathies. In the perspective of future clinical trials, reaching out a confirmed genetic and histopathologic diagnosis and establish internationally accepted care measures are pivotal to reduce the disease burden of congenital myopathies patients.
The core clinical features of congenital myopathies are early onset hypotonia, delayed motor milestones, muscular weakness, and respiratory muscle involvement1. There is wide variability in severity. The most severe forms show profound muscle weakness or fetal akinesia, bulbar weakness with deglutition problems and drooling, and respiratory involvement necessitating assistance. Patients can show dysmorphic features or skeletal deformities or arthrogryposis. Polyhydramnios or reduced fetal movements may be present during pregnancy. Miscarriages are often reported by patients’ mother in cases of autosomal recessive inheritance.
The classic phenotype associate neonatal hypotonia, gross motor delay, and proximal and axial weakness showing different degrees of severity. Recurrent respiratory infections are often reported. Patients may show skeletal deformities such as pectus excavatum, flat thorax and they develop scoliosis during growth spurt. At physical examination, patients have thin muscle bulk and reduced body weight. Facial and maxillary alterations including high-arched palate, tubular nose or low set ears are frequent and, in some conditions, there is a high-pitched or nasal voice. Ocular involvement including ptosis and ophtalmoplegia is present in specific congenital myopathies2.
Muscle weakness is mainly proximal and axial and patients report frequent falls, neck weakness or difficulties rising up from the supine position, necessitating rolling on one side. Facial weakness is recognizable with patients showing open mouth with an inverted V shape. Some congenital myopathies manifest with distal muscle involvement of legs and foot drop3. In the milder forms, difficulties in sport activities or effort intolerance can be the only manifesting signs, with patients developing muscle weakness later in life.
In general, patients tend to ameliorate during infancy and adolescence. However, certain forms are particularly aggressive leading to weakness progression and early adulthood gait loss (e.g. reducing body myopathy, centronuclear myopathies).
Some patients show prominent fatigability, effort intolerance or fluctuations that are due to the presence of secondary alterations of the neuromuscular junction4. Myalgias are frequent in patients with ‘core myopathies’ linked to RYR1 gene mutations (see below).
Serum creatine kinase (sCK) level is normal or slightly elevated. Electrophysiological studies and electromyography show myopathic features.
Whole body muscle MRI is very useful in orientating the genetic screening. In fact, different profiles of distribution of muscle alterations have been found as constant features in particular entities5.
Muscle biopsy analysis with both light and electron microscopy techniques is a useful tool to confirm the diagnosis and orientate genetic analyses. However, these analyses must be performed in highly skilled and experienced centers in order to allow a good interpretation and avoid technical artifacts1. Histopathologic features of each condition will be described in the sections relative to specific disorders.
Genetic pedigree and a careful family history are fundamental to establish the possible pattern of inheritance and start genetic analyses. More and more genes associated to congenital myopathies are described thank to exome/whole exome sequencing approaches6. For this reason, a constant and integrated crosstalk between clinicians, histopathologists, and geneticists is necessary to provide the best accurate diagnosis for every patient. Nevertheless around 50 percent of patients do not show mutations in the already know genes. We expect to identify novel congenital myopathies in the next years.
A multidisciplinary approach is necessary for the follow up of congenital myopathy patients. This should include the intervention of pediatric neurologist, orthopedic, pulmonologist, cardiologist, nutritionist, and physical therapist. Clinician should refer the guidelines published by the International Standard of Care Committee for Congenital Myopathies’ group7.
SPECIFIC ENTITIESNemaline myopathiesNemaline (from the Greek word néma=rod) or rod myopathies (NM) probably represent the congenital myopathy most frequently encountered. They are characterized by the presence of small, rod-shaped inclusions inside the cytoplasm of the fiber8. Clinical spectrum is wide and comprises forms manifesting antenatally, or milder form with childhood onset weakness. NM are genetically heterogeneous and inheritance can be autosomal dominant (AD), de novo, or autosomal recessive. This heterogeneity is demonstrated by the fact that twelve genes:
- 1.
ACTA1, MIM#161800
- 2.
NEB, MIM#256030
- 3.
TPM2, MIM#609285
- 4.
TPM3, MIM#609284
- 5.
TNNT1, MIM#605355
- 6.
KBTBD13, MIM#609273
- 7.
CFL2, MIM#610687
- 8.
KLHL40 MIM#615340
- 9.
KLHL41 MIM#615731
- 10.
LMOD3 MIM#616112
- 11.
MYO18B MIM# 607295
- 12.
MYPN MIM#608517
Have been associated with these conditions8,9. The most frequently mutated gene is the nebuline gene, NEB, in around 50 percent of the genetically identified forms10. NM linked to actin gene, ACTA1, is the second most frequent form with more than 200 mutations reported11. Cases with ACTA1 de novo mutations seem to be particularly severe, while dominant forms show a classic nemaline myopathy phenotype.
Cardiac involvement is not considered a core clinical feature of nemaline myopathies. However, ACTA1 NM, the recently described forms linked to MYOB18, myosin 18B, coding for a non-conventional myosin, and MYPN, myopalladin, a Z-line protein, can associate both myopathy and cardiomyopathy12–9.
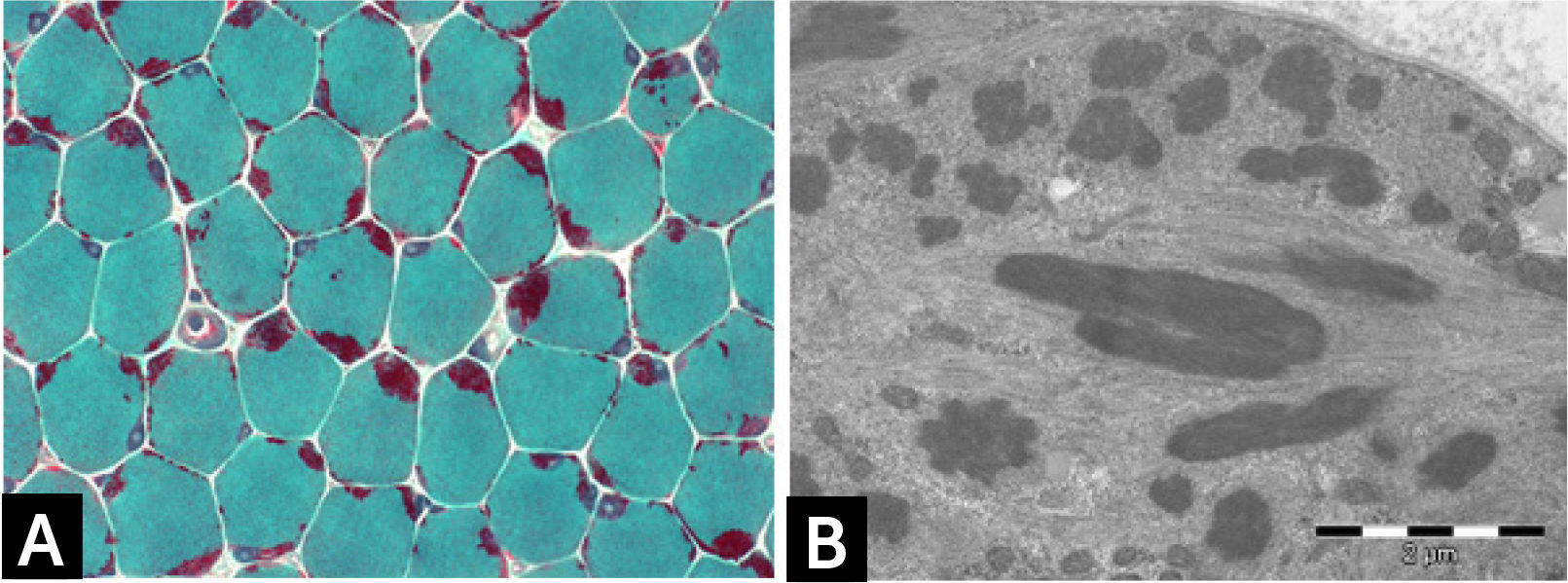
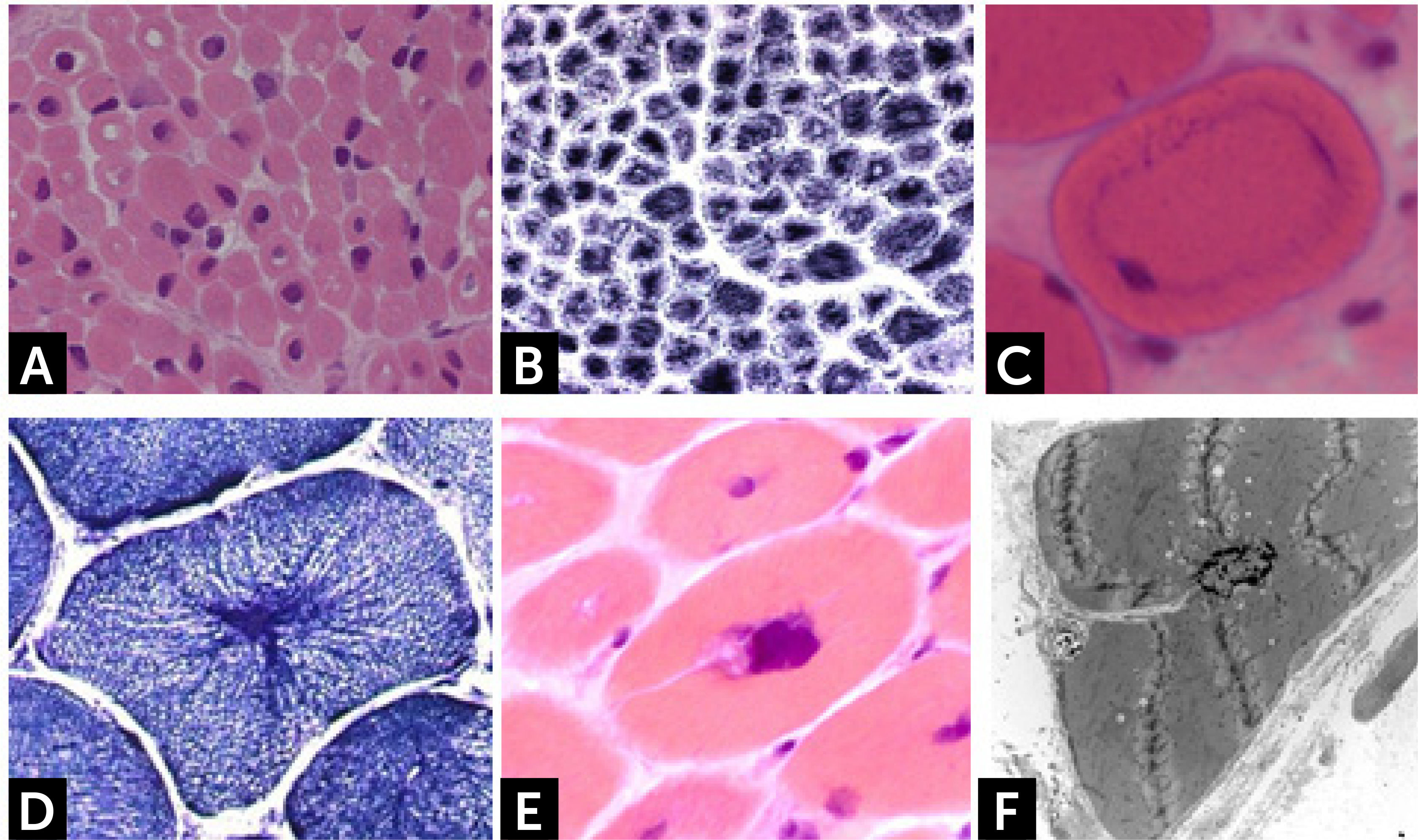
Histologically, all nemaline myopathies show the presence of fuchsinofilic protein inclusions visible with Gömöri trichrome staining (Fig. 1A). The latter are often elongated, and vary in number and distribution inside the muscle fiber. In newborn muscle biopsies, rods identification can be particularly tricky. In these cases, electron microscopy is mandatory in order to disclose the presence of rods. With this technique they appear as electron dense bodies measuring 1–7μm length and 0.3-2μm width (Fig. 1B). They present an orthogonally organized filamentous structure resembling to Z-line material. The exact origin and mechanisms of rod formation are largely unknown. Rods can be found inside myonuclei in ACTA1 and MYOPN NM11–9.
CONGENITAL MYOPAHIES WITH CORES![] Nemaline myopathy with mutations in NEB gene; (A) Clusters of rods at the periphery of fibers staining red at GT. (B) Electron micrograph showing nemaline bodies transversally and longitudinally oriented.](https://static.elsevier.es/multimedia/07168640/0000002900000006/v2_201901090613/S0716864018301226/v2_201901090613/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTzQ+NkT67GW3k0bS09oM5XwIBS1nAO29lrKzv7upGYpyZkvjb8/cXFkf3PaRzyggoKUUiQWA4ANLcYWXTIk4sgV6jCCN4LAp9SX5oU7sB/UEee/gMhLX/qXvRVrnh81e4pkV2NpW/giOdtbT5GJmG0p9qe3oQGu+3earsAfzir9diMBBeTvR3yfkd4xuHfiLUxFkNzKIzCNpJejyYkXC0j3 "] Nemaline myopathy with mutations in NEB gene; (A) Clusters of rods at the periphery of fibers staining red at GT. (B) Electron micrograph showing nemaline bodies transversally and longitudinally oriented.")
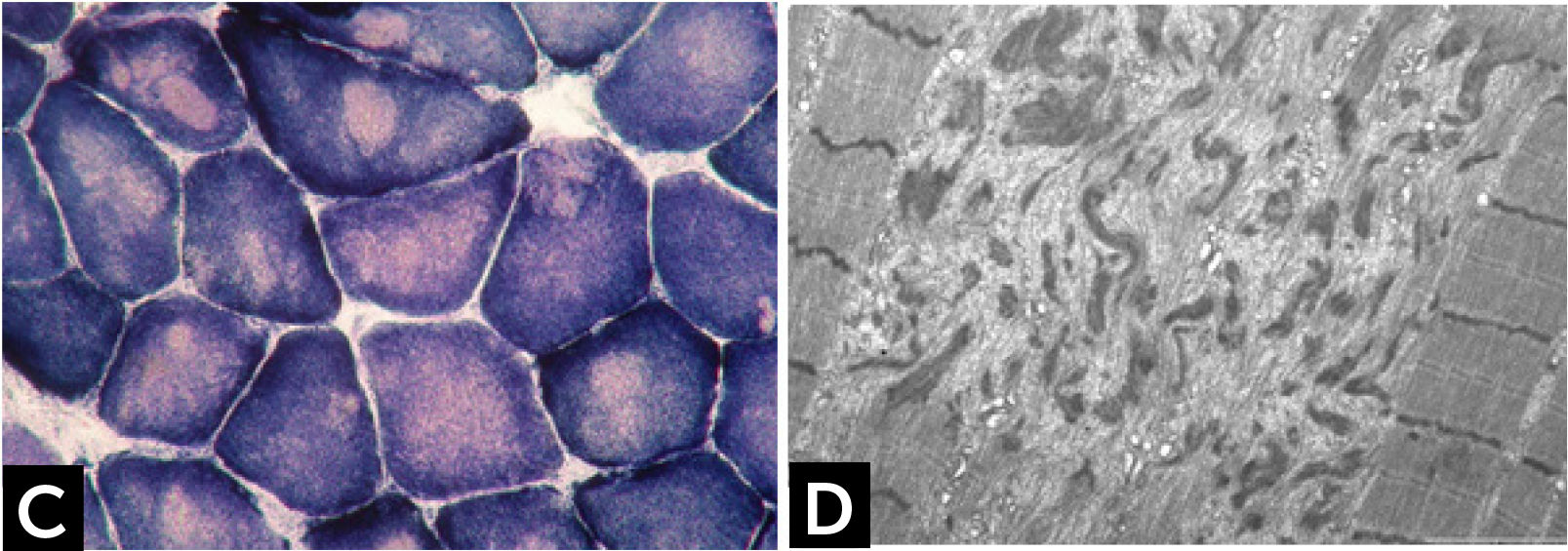
Core lesions are defined as well-defined rounded areas lacking oxidative activity (SDH, NADH etc.) corresponding to myofibrillar disorganization (Fig. 1C). By electron microscopy a typical core correspond to wide areas of compacted and disorganized myofibrils, with Z-line streaming and absence of mitochondria, extending over numerous sarcomeres or almost along the full length of the fiber (Fig. 1D)13. Cores can be central and single, single and peripheral, or multiple. Patients with core myopathies can present profound clinical severity with antennal manifestation or develop a proximal and mild muscle weakness in childhood or early adulthood. Inheritance is autosomal dominant or recessive and the majority of patients show mutation(s) of the RYR1 gene, coding for the skeletal muscle ryanodin channel receptor, which is also associated with susceptibility to malignant hyperthermia and rhabdomyolysis14. AR RYR1 mutated patients can show ocular involvement with ptosis and ophtalmoplegia. AD mutations in MYH7 or CCD18 genes have been also associated with ‘core’ myopathies15,16.
![] Central core disease. (C) Centrally located cores evident as well-defined areas devoid of NADH-TR activity. (D) Electron micrograph of a core showing contraction of myofibrils and disruption of the Z-line.](https://static.elsevier.es/multimedia/07168640/0000002900000006/v2_201901090613/S0716864018301226/v2_201901090613/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTzQ+NkT67GW3k0bS09oM5XwIBS1nAO29lrKzv7upGYpyZkvjb8/cXFkf3PaRzyggoKUUiQWA4ANLcYWXTIk4sgV6jCCN4LAp9SX5oU7sB/UEee/gMhLX/qXvRVrnh81e4pkV2NpW/giOdtbT5GJmG0p9qe3oQGu+3earsAfzir9diMBBeTvR3yfkd4xuHfiLUxFkNzKIzCNpJejyYkXC0j3 "] Central core disease. (C) Centrally located cores evident as well-defined areas devoid of NADH-TR activity. (D) Electron micrograph of a core showing contraction of myofibrils and disruption of the Z-line.")
The minicores are characterized by the presence of multiple foci of sarcomeric disorganization, with Z-line streaming, running over a few sarcomeres, even if occasionally they are longer with poorly-defined borders; mitochondria are absent from the altered areas17. Patients with minicores myopathy linked to AR mutations in selenoprotein N, SEPN1 gene present with a constant clinical phenotype characterized by hypotonia and gross motor delay followed by prominent axial and diaphragmatic weakness associated with rigid spine, scoliosis, and respiratory failure18. Some patients showing ptosis, ophtalmoplegia and severe muscle weakness harbor AR mutations in the RYR1 gene19,20. Titin gene (TTN) mutations have been associated with congenital myopathy with contractures, possible cardiac involvement and cardiomyopathy and minicores in muscle biopsy21,22.
Core-rod myopathyCore-rod myopathy shows the association of nemaline bodies with well-defined cores within separate muscle fiber regions (Fig. 1E). Numerous core-rod myopathies are associated with autosomal dominant (AD) or autosomal recessive (AR) mutations in the gene encoding the skeletal muscle ryanodine receptor (RYR1)23,24. In contrast, only few clinically heterogeneous patients with core-rod myopathy linked to nebulin (NEB) gene mutation have been reported. Clinical features can correspond to typical congenital phenotype or consist of prominent distal weakness with bilateral foot-drop3.
Reducing body myopathy![] Core-rod myopathy. Electron micrographs showing a fiber harboring both rods (indicated by a yellow star) and a core lesion (indicated by a blue arrow).](https://static.elsevier.es/multimedia/07168640/0000002900000006/v2_201901090613/S0716864018301226/v2_201901090613/es/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTzQ+NkT67GW3k0bS09oM5XwIBS1nAO29lrKzv7upGYpyZkvjb8/cXFkf3PaRzyggoKUUiQWA4ANLcYWXTIk4sgV6jCCN4LAp9SX5oU7sB/UEee/gMhLX/qXvRVrnh81e4pkV2NpW/giOdtbT5GJmG0p9qe3oQGu+3earsAfzir9diMBBeTvR3yfkd4xuHfiLUxFkNzKIzCNpJejyYkXC0j3 "] Core-rod myopathy. Electron micrographs showing a fiber harboring both rods (indicated by a yellow star) and a core lesion (indicated by a blue arrow).")
Reducing bodies were described around 40 years ago as cytoplasmic inclusions that reduce nitro-blue tetrazolium (NBT) and thus stain strongly with the menadione-NBT reaction25. Pioneer studies speculated that reducing activity of the bodies is likely to be due to protein sulfhydryl groups. More recently, it has been demonstrated that RBs correspond to protein aggregates whose major constituent is FHL126. Mutations in FHL1 gene (OMIM 300163) have been associated with reducing body myopathy (RBM), and other disorders26.
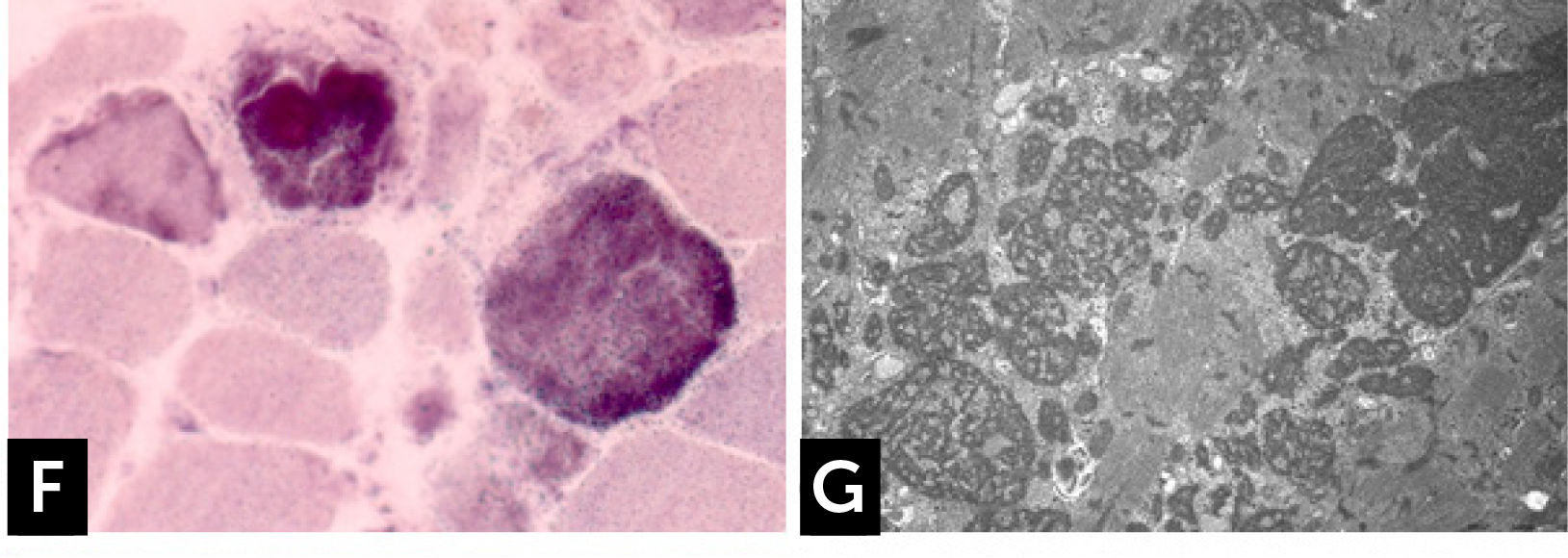
The clinical phenotype varies from severe, congenital or early childhood onset disorders manifested with delayed motor milestones, prominent muscle weakness and rapid loss of ambulation, rigid spine, to milder, less progressive conditions manifested later in life27. RBs are observed as bright pink cytoplasmic inclusions on H&E and red on the modified Gömöri trichrome stain. They strongly react for menadione-NTB with and without alpha-glycerolphosphate, (Fig. 1F) but they are devoid of oxidative, especially SDH and COX, and ATPase activities. The number and the size of RBs can vary among the samples and some fibers are completely replaced by them. Cytoplasmic bodies are seen as collections of red granules in the vicinity of reducing bodies. By electron microscopy, RBs correspond to large inclusions composed by osmiophilic granular material (Fig. 1G). Immuno-electron microscopy studies showed that FHL1 is indeed found inside the RB and confirmed the expected FHL1 localization in skeletal muscle adjacent to the Z-line/band I28.
Cap myopathy![] Reducing body myopathy (RBM) due to FHL1 mutations. (F) RBs stain blue strongly reducing NBT with menadione reaction without α-glycerophosphate. (G) Electron microscopy. RB material corresponds to coarse tubulofilaments.](https://static.elsevier.es/multimedia/07168640/0000002900000006/v2_201901090613/S0716864018301226/v2_201901090613/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTzQ+NkT67GW3k0bS09oM5XwIBS1nAO29lrKzv7upGYpyZkvjb8/cXFkf3PaRzyggoKUUiQWA4ANLcYWXTIk4sgV6jCCN4LAp9SX5oU7sB/UEee/gMhLX/qXvRVrnh81e4pkV2NpW/giOdtbT5GJmG0p9qe3oQGu+3earsAfzir9diMBBeTvR3yfkd4xuHfiLUxFkNzKIzCNpJejyYkXC0j3 "] Reducing body myopathy (RBM) due to FHL1 mutations. (F) RBs stain blue strongly reducing NBT with menadione reaction without α-glycerophosphate. (G) Electron microscopy. RB material corresponds to coarse tubulofilaments.")
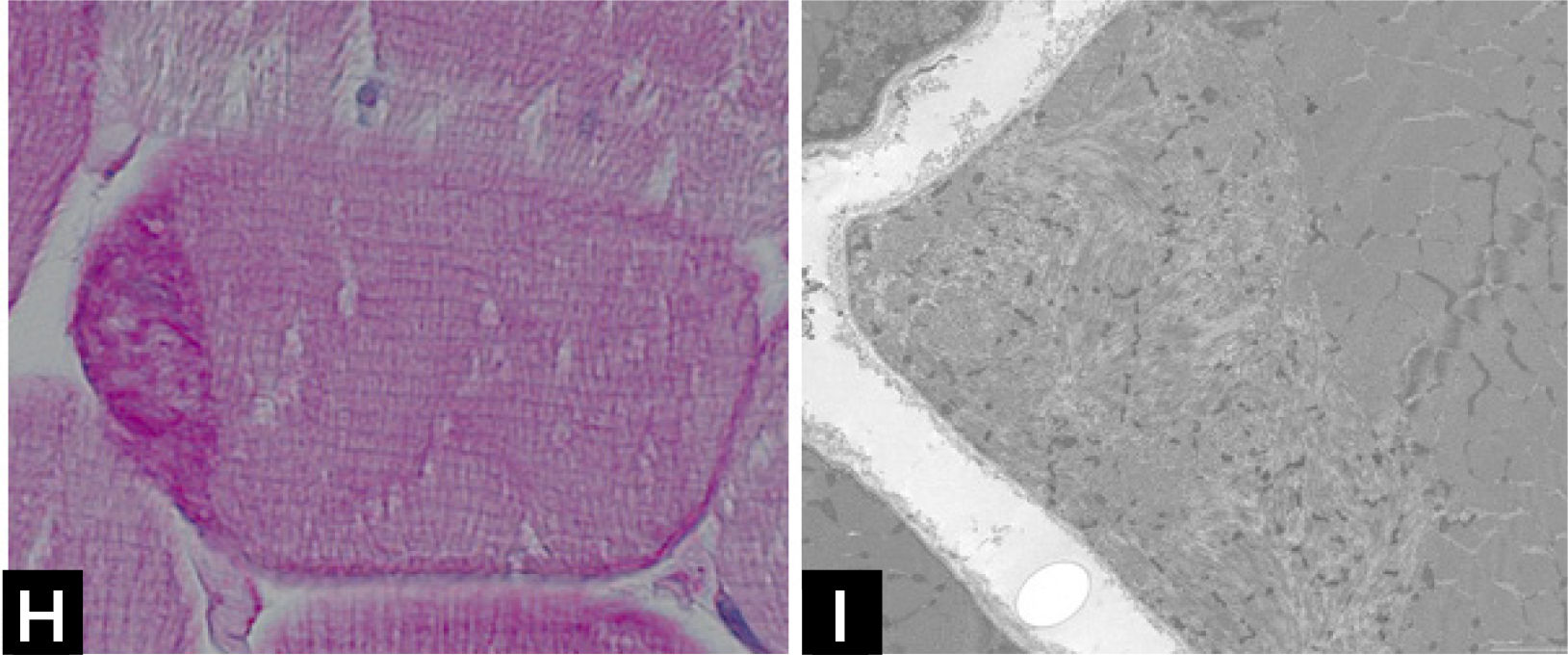
Cap myopathy is a rare congenital myopathy, characterized by the presence of peripherally-placed, well-delimited structures resembling a “cap”29. Clinical features are variable with forms presenting like a typical congenital myopathies and others showing prominent respiratory involvement and marked maxillofacial deformations1. Caps appear green and granular with Gömöri trichrome, stain strongly with PAS (Fig. 1H), and NADH, but they are pale with SDH. ATPases reactions do not stain the cap structures.
![] Cap disease; (H) Cap structure appearing as well-demarcated polar structures with intense PAS staining. (I) Electron micrograph of a Cap showing amorphous material with dispersed Z-line fragments.](https://static.elsevier.es/multimedia/07168640/0000002900000006/v2_201901090613/S0716864018301226/v2_201901090613/es/main.assets/gr5.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd2Vt2E9KIXSbfPNY5VCUB4kpCjPTZRm5n9r2Wgu2xKnaaLdMEH3EdygauzKlTyPTzQ+NkT67GW3k0bS09oM5XwIBS1nAO29lrKzv7upGYpyZkvjb8/cXFkf3PaRzyggoKUUiQWA4ANLcYWXTIk4sgV6jCCN4LAp9SX5oU7sB/UEee/gMhLX/qXvRVrnh81e4pkV2NpW/giOdtbT5GJmG0p9qe3oQGu+3earsAfzir9diMBBeTvR3yfkd4xuHfiLUxFkNzKIzCNpJejyYkXC0j3 "] Cap disease; (H) Cap structure appearing as well-demarcated polar structures with intense PAS staining. (I) Electron micrograph of a Cap showing amorphous material with dispersed Z-line fragments.")
Electron microscopy confirms the presence of well-delimited structures containing fragments of sarcomeres, cellular debris, and amorphous material (Fig. 1I). Mutations have been identified in the tropomyosin-2 (TPM2), in the tropomyosin-3 (TPM3), and more recently in the alpha-actin gene (ACTA1)2. The association of caps and nemaline bodies in the same patient have been described by us in TPM3-related cap myopathy29. We recently described a cohort of patients sharing a slowly progressive congenital cap myopathy with facial involvement and asymmetric weakness, and homozygous truncating mutations in MYPN gene, encoding myopalladin30.
Centronuclear myopathiesCentronuclear congenital myopathies are genetic conditions characterized by the presence of a high incidence of centrally or internally placed nuclei in rows in muscle fibers31. Four forms have identified: (1) a severe/lethal X-linked recessive form, also named myotubular myopathy with mutation in the MTM1 gene; (2) a clinically variable sporadic or AD form associated with DNM2 gene mutation, and (3) a moderate or severe AR or AD form related to BIN1 gene mutations. (4). Recently AR mutations in SPEG gene have been found in three patients presenting severe clinical form of congenital myopathy with dilated cardiomyopathy32.
Muscle biopsies of boys presenting the X-linked MTM1 form show the presence of centrally located nuclei (Fig. 2A), and a subsarcolemmal pale halo evident with oxidative enzymes (Fig. 2B). Female carriers of one MTM1 mutation may show mild muscle weakness and present a histopathologic findings consisting of basophilic loops found in the muscle fibers cytoplasm (Fig. 2C). These structures resemble a necklace and show increased oxidative activity and PAS staining31.
 H&E staining of frozen muscle section from a patient presenting myotubular myopathy (MTM1). Presence of rounded muscle fibers with centrally located nuclei, resembling myotubes. (B) NADH reaction showing clear peripheral halo. (C) Necklace fiber in a biopsy from a female MTM1 symptomatic carrier. (D) Particular radiated disposition of the sarcoplasmic reticulum as the “spokes of a wheel” in a DNM2 mutated patient muscle. (E) Cluster of myonuclei and membrane invagination in a muscle biopsy from a patient with AR BIN1 mutations. (F) Electron microscopy shows a centralized nucleus with membrane invagination.")
(A) H&E staining of frozen muscle section from a patient presenting myotubular myopathy (MTM1). Presence of rounded muscle fibers with centrally located nuclei, resembling myotubes. (B) NADH reaction showing clear peripheral halo. (C) Necklace fiber in a biopsy from a female MTM1 symptomatic carrier. (D) Particular radiated disposition of the sarcoplasmic reticulum as the “spokes of a wheel” in a DNM2 mutated patient muscle. (E) Cluster of myonuclei and membrane invagination in a muscle biopsy from a patient with AR BIN1 mutations. (F) Electron microscopy shows a centralized nucleus with membrane invagination.
Dynamin-2 (DNM2) gene, encodes for a large GTPase implicated in endocytosis and membrane trafficking. DNM2 mutations are associated both to autosomal dominant (AD) forms of CNM (mild or late-childhood and adult onset forms) and sporadic cases (severe early childhood type). Muscle biopsies show a high percentage of nuclear internalizations located especially in small hypotrophic fibers. Numerous fibers show the “radiated sacoplasmic strands” (RSS fibers), that correspond to a particular radiated disposition of the sarcoplasmic reticulum giving to the fiber the aspect of a bicycle wheel (Fig. 2D). Mutations in the amphiphysin 2 (BIN1) gene can be found both at AR state in severe neonatal or early-childhood type or at AD33-35 in the adult-onset progressive myopathy. Clusters of centrally located nuclei are found in a homogeneous population of rounded atrophic type 1 fibers. Connective tissue augmentation and fibro-adipose replacement without necrosis and regeneration are also seen. The presence of clusters of centrally located nuclei (Fig. 2E) surrounded by abundant amorphous material containing autophagic vacuoles and sarcolemmal membrane alterations consisting of invaginations, (Fig. 2F), vacuolization, and triads alterations are observed by electron microscopy (personal observation)34.
Congenital fiber-type disproportion (CFTD)CFTD is defined by the presence of smaller type 1 fibers (at least 12%) compared to type 2 fibers. Such change has been regarded for a long time as unspecific due to its presence in numerous congenital myopathies. Patients usually present with neonatal hypotonia variably associated with contractures. More recently, mutations in TPM3, TPM2, RYR1, ACTA1, SEPN1, and MYH7 genes have been found in congenital myopathy patients showing CFTD without any other histopathological lesion2.
DISEASES MECHANISMSThe mechanisms leading to muscle fiber degeneration and muscle weakness in congenital myopathies are largely unknown. Regarding the group of nemaline myopathies, all the known defective protein in these conditions are sarcormeric structural protein such as nebulin, actin, tropomyosins or myopalladin, thin filaments Kelch associated proteins, or the non-conventional myosin 18 B protein, found in the proximity of the Z line. All this proteins belong to the sarcomere, the constitutive myofibrillar unit necessary for muscle contraction. We can compare the sarcomere as a highly organized structure resembling the pentagram of a music partition. Thus, it is possible that a structural sarcomeric modification provoked by a genetic alteration could lead to its disarray. In the case of nemaline myopathies the sarcomeric derangement could lead to nemaline bodies accumulation.
Fine morphological studied conducted by us on muscle samples from patients with NEB mutations showed a positive correlation between clinical severity and myofibrillar smallness and disruption associated with rods deposition in severe/lethal cases10. Other studies measuring single fibers contractility in muscle biopsies from patients with different nemaline myopathies confirmed that the contractile alteration is related to the shortness of thin filaments34.
In the case of core myopathies linked to RYR1 gene mutations, the possible pathogenic mechanisms could be related to calcium mishandling/toxicity provoked by altered RYR1 channel function14. However, the relation between calcium mishandling and core lesion formation remains to be clarified.
In the centronuclear myopathy group, a mechanism of altered development of skeletal muscle fibers have been hypothesized in the myotubular myopathy, while DNM2 and amphysisin 2 seem to be involved in the intracellular trafficking31.
CONCLUSIONSIn this article, we reviewed main clinical, morphological and genetic features of the main congenital myopathies. It is possible that other congenital myopathies forms will be discovered in future years with the application of more advanced sequencing approaches such as RNA sequencing. The future challenge for clinicians, researchers and patients will be the development of therapeutic approach. With this in mind, a deep clinical, morphological, and genetic phenotyping are and remain essential steps for the understanding of this group or myopathies.
Declaration of interestThe author has nothing to disclose.