






Las miopatías metabólicas son enfermedades que afectan el músculo esquelético. Son causadas por deficiencias enzimáticas genéticamente determinadas, que afectan el metabolismo del glicógeno, de los lípidos, de la cadena respiratoria mitocondrial o de las purinas.
Las manifestaciones clínicas son variables desde síntomas permanentes como la debilidad progresiva; síntomas episódicos como la intolerancia al ejercicio, mialgias y/o mioglobinuria; o en la combinación de síntomas episódicos y permanentes.
En este espectro de síntomas, la insuficiencia respiratoria y la insuficiencia renal aguda producto de una rabdomiólisis masiva representan los cuadros de mayor severidad.
El diagnóstico de este grupo heterogéneo de enfermedades se basa en la historia clínica, con la búsqueda dirigida de síntomas y potenciales desencadenantes de ellos, así como en la historia familiar en busca de consanguinidad y/o de síntomas relacionados en otros miembros de la familia.
El examen clínico en busca de debilidad generalizada o localizada (compromiso selectivo de algunos grupos musculares), hipertrofia muscular, o anormalidades en la relajación del músculo pueden orientar, aunque la ausencia de hallazgos relevantes al examen no excluye la posibilidad de una miopatía metabólica.
La caracterización de síntomas, antecedentes familiares y hallazgos del examen guía el estudio, el cual incluye exámenes básicos como al creatinkinasa, búsqueda de rabdomiólisis, así como estudios de compromiso sistémico finalizando habitualmente en estudio genético molecular.
Desde el punto de vista terapéutico evitar desencadenantes, asegurar aporte de glucosa en situaciones de sobreexigencia metabólica al músculo, prevención y manejo e la insuficiencia renal asociada a rabdomiolisis y en algunos casos reemplazo de la enzima deficiente deben plantearse muy precozmente a fin de evitar mayor morbilidad.
Metabolic myopathies are diseases that involve the skeletal muscle. They are caused by genetically determined enzymatic deficiencies that affect the metabolism of glycogen, lipids, mitochondrial respiratory chain or purines.
The clinical manifestations are variable from permanent symptoms such as progressive weakness; episodic symptoms such as exercise intolerance, myalgia and/or myoglobinuria; or in the combination of episodic and permanent symptoms.
In this spectrum of symptoms, respiratory failure and acute renal failure due to massive rhabdomyolysis represent the most severe cases.
The diagnosis of this heterogeneous group of illness based on the clinical history, with the directed search of symptoms and potential triggers of them, as well as in the family history in search of consanguinity and/or of related symptoms in other members of the family.
The clinical examination in search of generalized or localized weakness (selective involvement of some muscle groups), muscle hypertrophy, or abnormalities in muscle relaxation may help; although the absence of findings relevant to the examination does not exclude the possibility of a metabolic myopathy.
The characterization of symptoms, family history and findings of the study guide the study, which includes basic tests such as creatinkinase, search for rhabdomyolysis, as well as systemic commitment studies usually ending in molecular genetic study.
From the therapeutic point of view, avoid triggers, ensure glucose intake in situations of metabolic over-exertion to the muscle, prevention and management of renal failure associated with rhabdomyolysis and in some cases replacement of the deficient enzyme should be considered very early in order to avoid greater morbidity.
Son un grupo de trastornos hereditarios causados por deficiencias enzimáticas que comprometen el sistema energético del músculo.
Aunque sus manifestaciones pueden ser muy variables, dependiendo de la edad de inicio y factores ambientales, pueden agruparse clínicamente en tres grandes grupos de manifestaciones clínicas:
- A.
Síntomas permanentes (debilidad muscular progresiva)
- B.
Síntomas episódicos o de disfunción muscular intermitente (intolerancia al ejercicio, mialgias y/o mioglobinuria)
- C.
Mixtos: síndromes en que coexisten ambos tipos de síntomas
Es necesario tener en consideración que patología endocrinológica, estados carenciales, drogas y otros cuadros pueden afectar el metabolismo muscular en forma secundaria.
La comprensión del metabolismo energético del músculo es esencial para el diagnóstico y tratamiento de las miopatías metabólicas.
La energía necesaria para la contracción muscular implica varias vías metabólicas. Durante el reposo la mayor parte de la energía proviene del ATP producto de la oxidación de ácidos grasos. Durante el ejercicio la proporción de energía derivada de hidratos de carbono y lípidos depende de su duración e intensidad y del estado físico del individuo1. El ejercicio intenso de corta duración requiere de una alta proporción de hidratos de carbono como sustrato energético, a diferencia del ejercicio prolongado, en que aumenta la utilización de ácidos grasos.
El glicógeno es metabolizado a lactato o piruvato, proporcionando energía durante una actividad de alta intensidad. En condiciones aerobias, el piruvato entra en el ciclo de Krebs para generar energía a través del metabolismo oxidativo.
Los ácidos grasos de cadena larga, principal fuente de energía durante ejercicio sostenido, son transportados a la mitocondria como ésteres de acilcarnitina, reacción catalizada por la carnitina palmitoil-transferasa (CPT), donde son oxidados a acetil-CoA y ATP2.
En ejercicios breves se utiliza la fosfocreatina y el ciclo de nucleótidos de purina. La fosfocreatina restaura el nivel de ATP desde el ADP, mediante la transferencia de un grupo fosforilo, reacción catalizada por la creatinkinasa (o creatinfosfokinasa)3.
CLASIFICACIÓNDesde el punto de vista etiopatogénico las miopatías metabólicas se han agrupado en4:
- 1.
Trastornos del metabolismo del glicógeno
- 2.
Trastornos del metabolismo de los lípidos
- 3.
Defectos de la cadena respiratoria mitocondrial
- 4.
Trastornos del metabolismo de purinas (deficiencia de mioadenilato deaminasa)
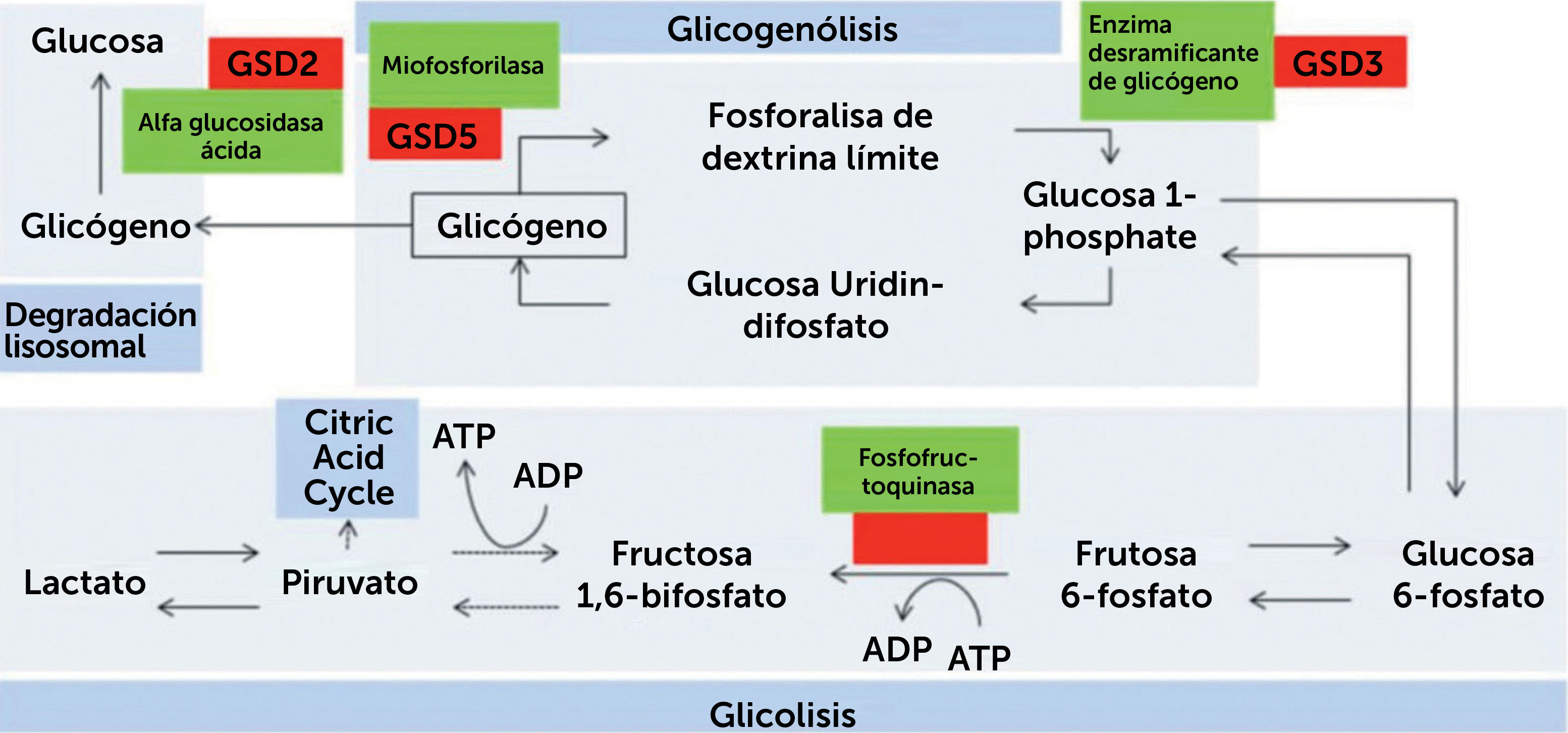
Las glicogenosis son trastornos que comprometen alguna de las vías metabólicas del glicógeno, ya sea síntesis de glicógeno (glicogenogénesis), degradación de glicógeno (glicogenolisis) o degradación de glucosa (glicólisis). Se denominan mediante la enzima específica deficitaria, un epónimo o números romanos ordenados según data de su identificación (Fig. 1).
. (Obtenido de: Lilleker JB, Keh YS, Roncaroli F, et al. Pract Neurol 2018;18:14–265)")
Resumen de vía metabólica del proceso de glicogenosis, glicólisis y degradación lisosomal
Se presentan enzimas involucradas en el proceso. Línea puntada representa los pasos intermedios omitidos. GSD: Trastorno de almacenamiento de glicógeno (de su sigla en inglés). (Obtenido de: Lilleker JB, Keh YS, Roncaroli F, et al. Pract Neurol 2018;18:14–265)
Las más frecuentes son el déficit de maltasa ácida (enfermedad de Pompe o Glicogenosis II) y el déficit de miofosforilasa (enfermedad de McArdle o Glicogenosis V).
Presentan variabilidad fenotípica y heterogeneidad genética, a nivel muscular se describen 2 síndromes clínicos: síndromes episódicos (intolerancia al ejercicio, calambres, mialgias y mioglobinuria recurrentes, como en tipo V) o síndromes fijos (hipotonía y debilidad muscular, como en tipo II). El compromiso de otros tejidos puede manifestarse como hipoglicemia, insuficiencia hepática, miocardiopatía o características específicas del compromiso mutisistémico.
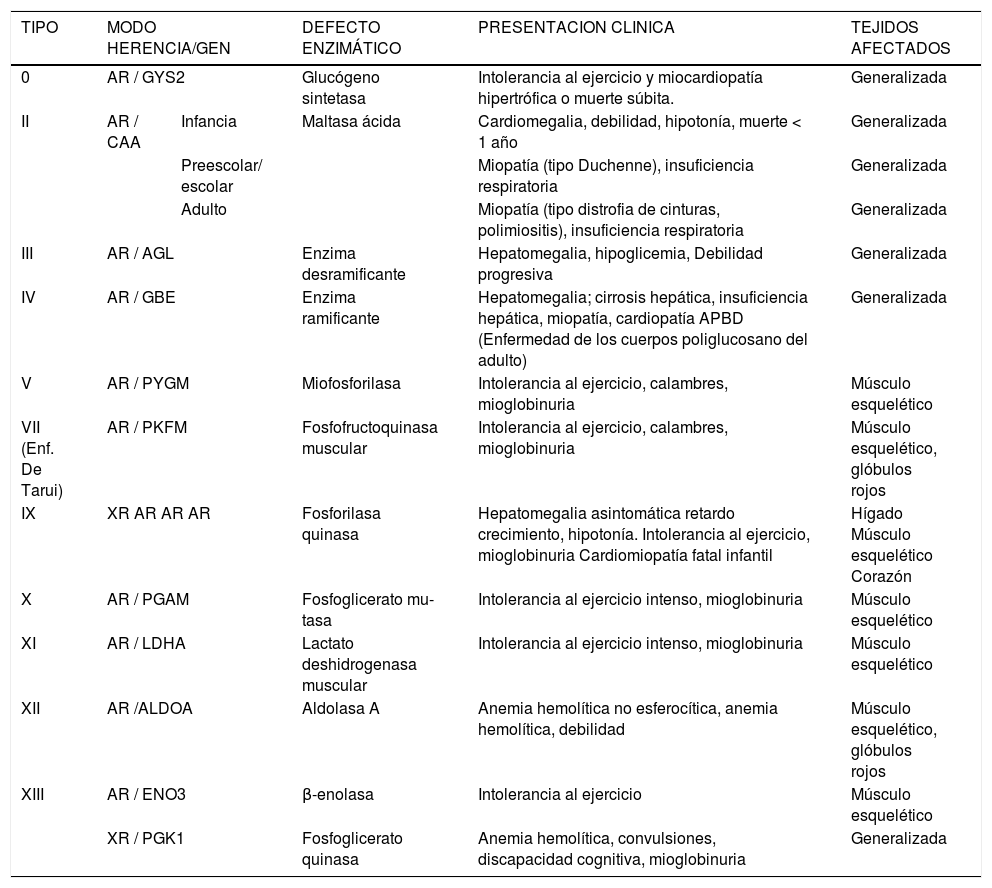
La Tabla 1 resume la distribución del tejido afectado, presentación clínica, defecto enzimático, y modo de herencia de las glicogenosis que comprometen principalmente el músculo.
Glicogenosis que afectan principalmente el músculo4
| TIPO | MODO HERENCIA/GEN | DEFECTO ENZIMÁTICO | PRESENTACION CLINICA | TEJIDOS AFECTADOS | |
|---|---|---|---|---|---|
| 0 | AR / GYS2 | Glucógeno sintetasa | Intolerancia al ejercicio y miocardiopatía hipertrófica o muerte súbita. | Generalizada | |
| II | AR / CAA | Infancia | Maltasa ácida | Cardiomegalia, debilidad, hipotonía, muerte < 1 año | Generalizada |
| Preescolar/ escolar | Miopatía (tipo Duchenne), insuficiencia respiratoria | Generalizada | |||
| Adulto | Miopatía (tipo distrofia de cinturas, polimiositis), insuficiencia respiratoria | Generalizada | |||
| III | AR / AGL | Enzima desramificante | Hepatomegalia, hipoglicemia, Debilidad progresiva | Generalizada | |
| IV | AR / GBE | Enzima ramificante | Hepatomegalia; cirrosis hepática, insuficiencia hepática, miopatía, cardiopatía APBD (Enfermedad de los cuerpos poliglucosano del adulto) | Generalizada | |
| V | AR / PYGM | Miofosforilasa | Intolerancia al ejercicio, calambres, mioglobinuria | Músculo esquelético | |
| VII (Enf. De Tarui) | AR / PKFM | Fosfofructoquinasa muscular | Intolerancia al ejercicio, calambres, mioglobinuria | Músculo esquelético, glóbulos rojos | |
| IX | XR AR AR AR | Fosforilasa quinasa | Hepatomegalia asintomática retardo crecimiento, hipotonía. Intolerancia al ejercicio, mioglobinuria Cardiomiopatía fatal infantil | Hígado Músculo esquelético Corazón | |
| X | AR / PGAM | Fosfoglicerato mu- tasa | Intolerancia al ejercicio intenso, mioglobinuria | Músculo esquelético | |
| XI | AR / LDHA | Lactato deshidrogenasa muscular | Intolerancia al ejercicio intenso, mioglobinuria | Músculo esquelético | |
| XII | AR /ALDOA | Aldolasa A | Anemia hemolítica no esferocítica, anemia hemolítica, debilidad | Músculo esquelético, glóbulos rojos | |
| XIII | AR / ENO3 | β-enolasa | Intolerancia al ejercicio | Músculo esquelético | |
| XR / PGK1 | Fosfoglicerato quinasa | Anemia hemolítica, convulsiones, discapacidad cognitiva, mioglobinuria | Generalizada | ||
Nomenclaturas: AR, autosómica recesiva; XR, ligado al X recesivo.
Puede manifestarse por intolerancia al ejercicio: cansancio, fatigabilidad, mialgias, calambres, debilidad, sensibilidad a la palpación muscular con relación a ejercicio, asociados en alrededor del 50% a mioglobinuria. Se inicia generalmente en edad escolar o adolescencia, y se ha estimado que afecta a 1 en 100000 personas6. Es un trastorno hereditario autosómico recesivo debido a más de 100 mutaciones diferentes del gen PYGM ubicado en el cromosoma 11q13.1 que codifica la miofosforilasa.
Durante largo tiempo se consideró una enfermedad clínicamente homogénea, sin embargo, sus manifestaciones pueden ir desde intolerancia al ejercicio leve hasta una condición invalidante, con calambres frecuentes y episodios recurrentes de mioglobinuria, en algunos casos acompañada de hipertrofia muscular (confundiéndose en ocasiones con la distrofia muscular de Becker)7.
El tipo y cantidad de ejercicio necesarios para precipitar los síntomas varía entre pacientes, sin embargo, lo más habitual es el ejercicio isométrico breve e intenso (anaer>bico). El ejercicio moderado es bien tolerado. Un fenómeno característico en pacientes con McArdle es el llamado fenómeno del segundo aire (“second wind phenomena”), consistente en el alivio de los síntomas (calambres dolorosos) al continuar con ejercicio de menor intensidad. La debilidad fija es excepcional en estos pacientes, pudiendo presentarse después de la 4a década de vida, como debilidad proximal.
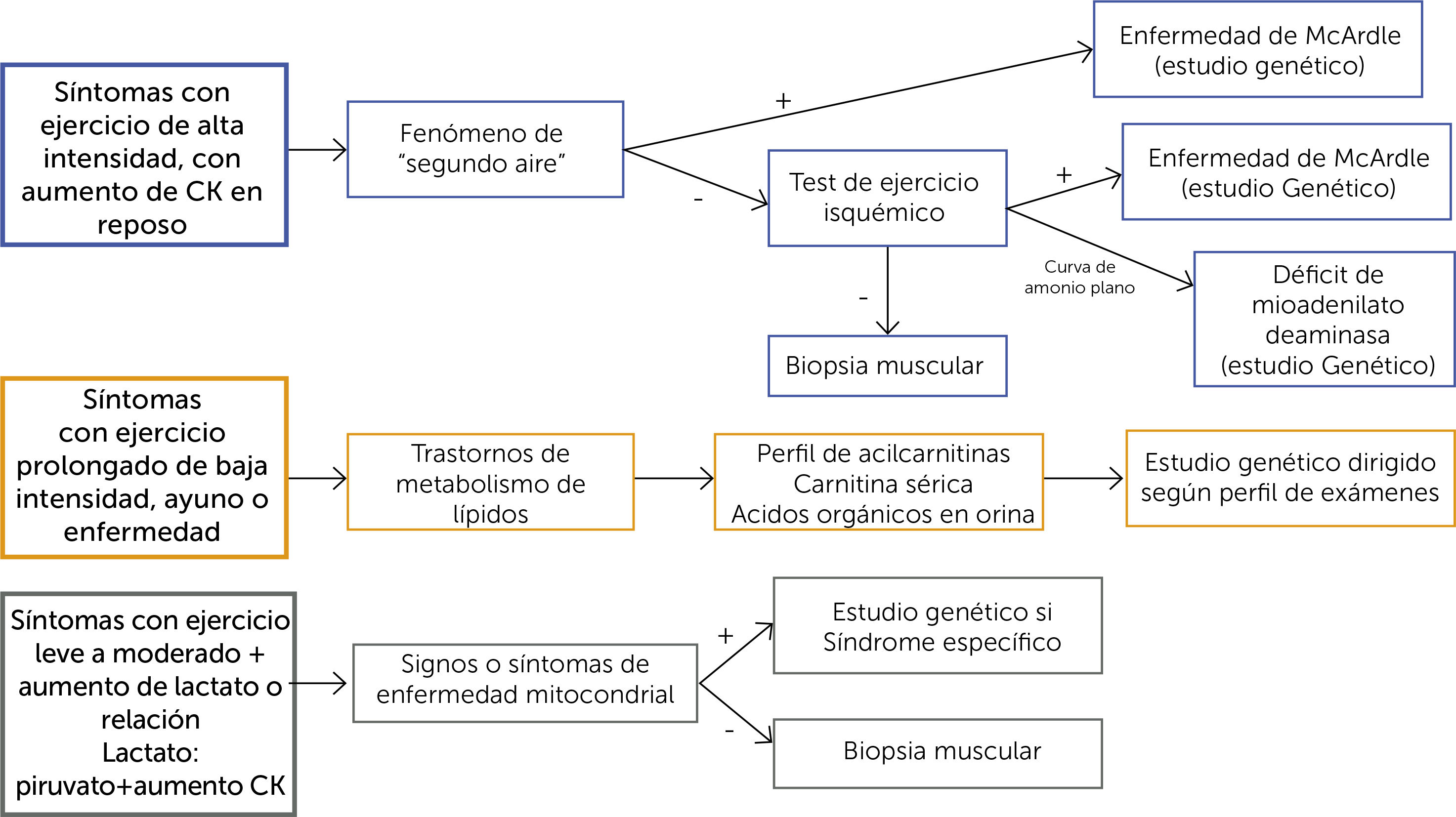
DiagnósticoSíntomas clínicos relacionados con ejercicio intenso o prolongado moderado, con calambres o mialgias, que afectan solo los músculos activados en el ejercicio.
Prueba de ejercicio isquémico: En individuos normales las curvas de lactato y amonio son ascendentes en el ejercicio. El lactato debe subir de 3 a 6 veces el valor basal. Una curva plana de lactato sugiere un defecto del metabolismo del glicógeno (McArdle). El amonio aumenta de 5 a 10 veces el valor basal, lo que confirma que la prueba es adecuada. CK sérica variablemente elevada (habitualmente mayor a 1000 U/I en reposo). El procedimiento se detalla en la tabla 2.
Prueba de ejercicio isquémico
| 1. Se recomienda paciente en ayuno de 8 horas y reposo absoluto los 30 min previos al inicio de la prueba. |
| 2. Se instala vía venosa permeable (calibre 20 G) en vena antecubital del antebrazo evitando estasia venosa en la zona. |
| 3. Se extrae muestra de sangre para medir lactato y amonio basales. |
| 4. Se instala manguito de presión de esfigmomanómetro manual en el brazo, habitualmente en la extremidad dominante. Se insufla aire hasta 20 mm de Hg sobre la presión sistólica, para asegurar la isquemia de la mano. |
| 5. Se solicita al paciente realización de ejercicio de flexo-extensión forzada de los dedos de la mano durante un minuto: apretar ergonómetro manual o una pelota, asegurando se mantenga la intensidad del ejercicio (máxima contracción voluntaria a un ritmo de 60 contracciones/min) hasta el agotamiento, sin detenerse. |
| 6. Se desinfla el manguito y se toman muestras de sangre venosa de láctico y amonio a los 1, 3, 5, 10 y 20 minutos finalizado el ejercicio. |
| 7. De cada una de las cinco extracciones de sangre se obtienen dos muestras de 3 ml (un tubo con heparina de sodio para el estudio del amonio y un tubo con fluorato sódico/oxalato potásico para el lactato) que se colocan en hielo y se envían a laboratorio en un tiempo inferior a los 30 min para centrifugación y posterior análisis. |
Biopsia muscular: ausencia de fosforilasa muscular y variable acumulación de glicógeno subsarcolemal, visible con reacción de PAS y en microscop-a electrónica.
Estudio de mutaciones en del gen PYGM: Actualmente se plantea que si la sospecha clínica es altamente sugerente de esta enfermedad y el test de ejercicio isquémico es categórico, se podría realizar directamente el estudio genético del gen PYGM y no realizarse la biopsia muscular.
Tratamiento
No hay hasta el momento tratamiento etiológico específico. Recomendaciones generales incluyen:
- -
Evitar situaciones que puedan precipitar mioglobinuria, como el ejercicio intenso.
- -
Alimentación alta en carbohidratos complejos y baja en grasa puede ser de utilidad para asegurar aporte de glucosa proveniente de las reservas de glicógeno hepáticas.
- -
En caso de ejercicio moderado y sostenido se recomienda evitar ayuno, e ingerir azúcar o sacarosa antes y durante el ejercicio.
- -
La suplementación con vitamina B6 60-90 mg/día (que juega un rol importante en la actividad enzimática de la fosforilasa muscular) ha mostrado mejoría en los síntomas de pacientes con actividad enzimática residual, mostrando aumento de los niveles enzimáticos en la biopsia muscular y aumento del lactato en el test de ejercicio isquémico8,9.
- -
Durante un episodio de mioglobinuria se recomienda hospitalización, optimizar hidratación, aporte de glucosa intravenosa, alcalinización de la orina y manejo analgésico con acetaminófeno ya que el uso de antinflamatorios no esteroidales (AINES) podría inducir crisis10.
Se considera la forma más grave y única glucogenosis con acumulación lisosomal y no citoplasmática de glicógeno. Es causada por déficit de la α-glucosidasa, enzima que cataliza la hidrólisis de glicógeno, lo que lleva acumulación de glicógeno en los tejidos, especialmente músculo esquelético, corazón e hígado. De herencia autosómica recesiva del gen GAA, existiendo más de 50 mutaciones identificadas. Su incidencia reportada en EEUU es de 1 en 40 mil nacimientos11.
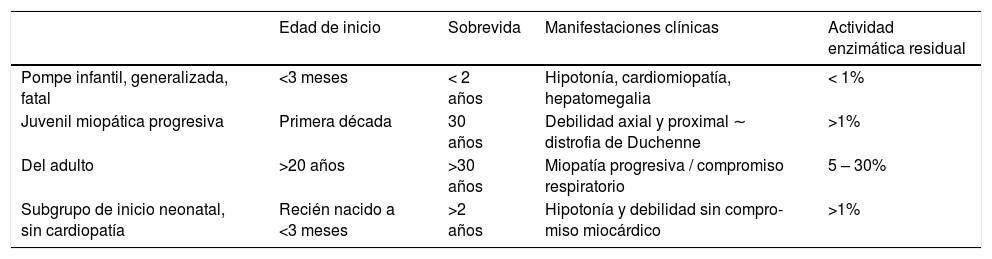
En la actualidad se plantea un espectro clínico continuo desde la forma infantil precoz fatal a la forma lentamente progresiva del adulto de inicio, incluso a edades muy avanzadas, siendo la severidad del cuadro determinada por la actividad enzimática residual. Las formas clásicas descritas se resumen en la Tabla 312.
Formas de presentación en Glucogenosis tipo II
| Edad de inicio | Sobrevida | Manifestaciones clínicas | Actividad enzimática residual | |
|---|---|---|---|---|
| Pompe infantil, generalizada, fatal | <3 meses | < 2 años | Hipotonía, cardiomiopatía, hepatomegalia | < 1% |
| Juvenil miopática progresiva | Primera década | 30 años | Debilidad axial y proximal ∼ distrofia de Duchenne | >1% |
| Del adulto | >20 años | >30 años | Miopatía progresiva / compromiso respiratorio | 5 – 30% |
| Subgrupo de inicio neonatal, sin cardiopatía | Recién nacido a <3 meses | >2 años | Hipotonía y debilidad sin compro- miso miocárdico | >1% |
se inicia en los primeros meses de vida con hipotonía y debilidad generalizada, miocardiopatía severa, macroglosia, trastorno de alimentación, retraso en desarrollo pondoestatural, e insuficiencia respiratoria. Sin tratamiento la sobrevida es limitada al primer año de vida a causa de insuficiencia cardiorrespiratoria progresiva13. La hipotonía puede estar presente desde el nacimiento o manifestarse después de las primeras semanas o meses de vida gatillados por una infección respiratoria. La debilidad en estos casos se explica por el compromiso directo del músculo y de la motoneurona de la asta anterior en la médula espinal. El cuadro clínico puede confundirse con una atrofia muscular espinal (AME) tipo I o enfermedad de Werdnig-Hoffmann.
El diagnóstico se basa en las características clínicas descritas. La electromiografía puede mostrar un patrón miopático y/o de denervación, dependiendo de la magnitud del compromiso de motoneurona asociado, y descargas pseudomiotónicas.
Glucogenosis Tipo II, forma juvenilLos síntomas aparecen entre los 2 y 5 años de edad siendo la cardiopatía inusual. Estos pacientes pueden manifestar retraso en adquisición de habilidades motoras con debilidad de cintura pélvica, con dificultad para subir escaleras, correr o pararse del suelo. La debilidad muscular es progresiva, de predominio en tronco y cintura pelviana, y destaca fatigabilidad y disnea en decúbito que mejora al incorporarse a sedente o pararse, ya que mejora la función diafragmática. Pueden desarrollar hiperlordosis, escoliosis, contracturas y dolor lumbar. Otras manifestaciones incluyen dificultad para masticar y tragar, síntomas respiratorios y apneas del sueño, manifestadas como cansancio, cefalea y somnolencia matinal, antes que la debilidad de extremidades sea evidente.
Por edad de presentación y manifestaciones clínicas debe considerarse en el diagnóstico diferencial de la distrofia muscular de Duchenne (DMD), distrofia muscular más frecuente en niños.
En algunos casos es posible encontrar hepatomegalia y macroglosia, pero rara vez cardiopatía significativa. La corrección quirúrgica de la escoliosis debe evaluarse cuidadosamente en relación con la función respiratoria, ya que las complicaciones respiratorias constituyen las causas principales de morbimortalidad. La sobrevida es variable y en la actualidad con la asistencia ventilatoria estos pacientes han logrado sobrevidas más prolongadas y mejor calidad de vida.
Glucogenosis Tipo II, forma de inicio tardíose caracteriza por debilidad muy lentamente progresiva de predominio proximal y axial14. Destaca la debilidad diafragmática, desproporcionada al grado de debilidad de extremidades, manifestándose con disnea y requerimientos de asistencia ventilatoria aún en las etapas en que se preserva la marcha, incluso la insuficiencia respiratoria puede presentarse como debut de la enfermedad, sin síntomas motores previos. Pueden presentar el llamado signo de Beevor, que corresponde al ascenso del ombligo al flectar el cuello estado en decúbito supino, signo que ha sido asociado clásicamente con la distrofia muscular facioescapulohumeral, pero que es inespecífico, pudiendo presentarse en otras enfermedades neuromusculares15.
Se describe en estas formas predilección por ciertos grupos musculares, tales como los músculos axiales, de cuello y abdominales, determinando en ocasiones mayor dificultad para la incorporación de decúbito a sedente y en la medida que avanza el compromiso axial, limitación de la movilidad espinal con espina rígida. Otros músculos que suelen comprometerse son: la musculatura bulbar con hipernasalidad, elevadores de párpados determinando ptosis a edades de aparición variables y disartria por compromiso de músculos linguales. El compromiso cardiaco es poco frecuente en las formas adultas, y cuando existe es menos significativo que en las formas infantiles o juveniles16.
Diagnóstico
Se establece basado en clínica, asociado a una CK que se encuentra elevada 2 a 10 veces sobre el valor normal en 90% de los casos. HiperCKemias sobre 100 veces el valor normal obligan a considerar otros diagnósticos. El hemograma puede evidenciar linfocitos con depósitos de glicógeno.
El estudio cardiológico y de función pulmonar debe efectuarse de regla en todos los pacientes. En esta enfermedad la espirometría en posición de decúbito puede mostrar mayor compromiso ventilatorio que en posición sedente, siendo una caída en la capacidad vital forzada mayor al 20% indicación de asistencia ventilatoria.
Las imágenes por resonancia de músculo pueden mostrar un compromiso precoz del aductor magnus y recto femoral incluso en pacientes asintomáticos17.
La biopsia muscular puede evidenciar una miopatía vacuolar PAS-positiva con distorsión completa del patrón miofibrilar o cambios miopáticos mínimos y en la microscop-a electrónica acúmulos de glicógeno lisosomal. En algunos casos puede no ser concluyente.
El estudio enzimático para maltasa ácida en sangre seca (papel filtro) o fresca es el primer estudio que nos establece el diagnóstico, y es considerado de primera línea en la actualidad si existe la sospecha clínica.
Si el estudio enzimático en sangre es positivo, este debe ser confirmado con la secuenciación del gen GAA o con estudio enzimático en tejidos (músculo o fibroblastos)18.
Tratamiento
La terapia de reemplazo enzimático con α-glucosidasa intravenosa recombinante es la única terapia eficaz en esta enfermedad, en todas las edades. La mayoría de los recién nacidos tratados muestran prolongación de sobrevida, evolucionando con mejor desarrollo motor y función miocárdica. Se describen cambios en la función e histología del músculo y en función respiratoria en formas de inicio tardío19,20. También es necesario monitorizar la aparición de trastornos ventilatorios, escoliosis y osteoporosis, en el seguimiento21.
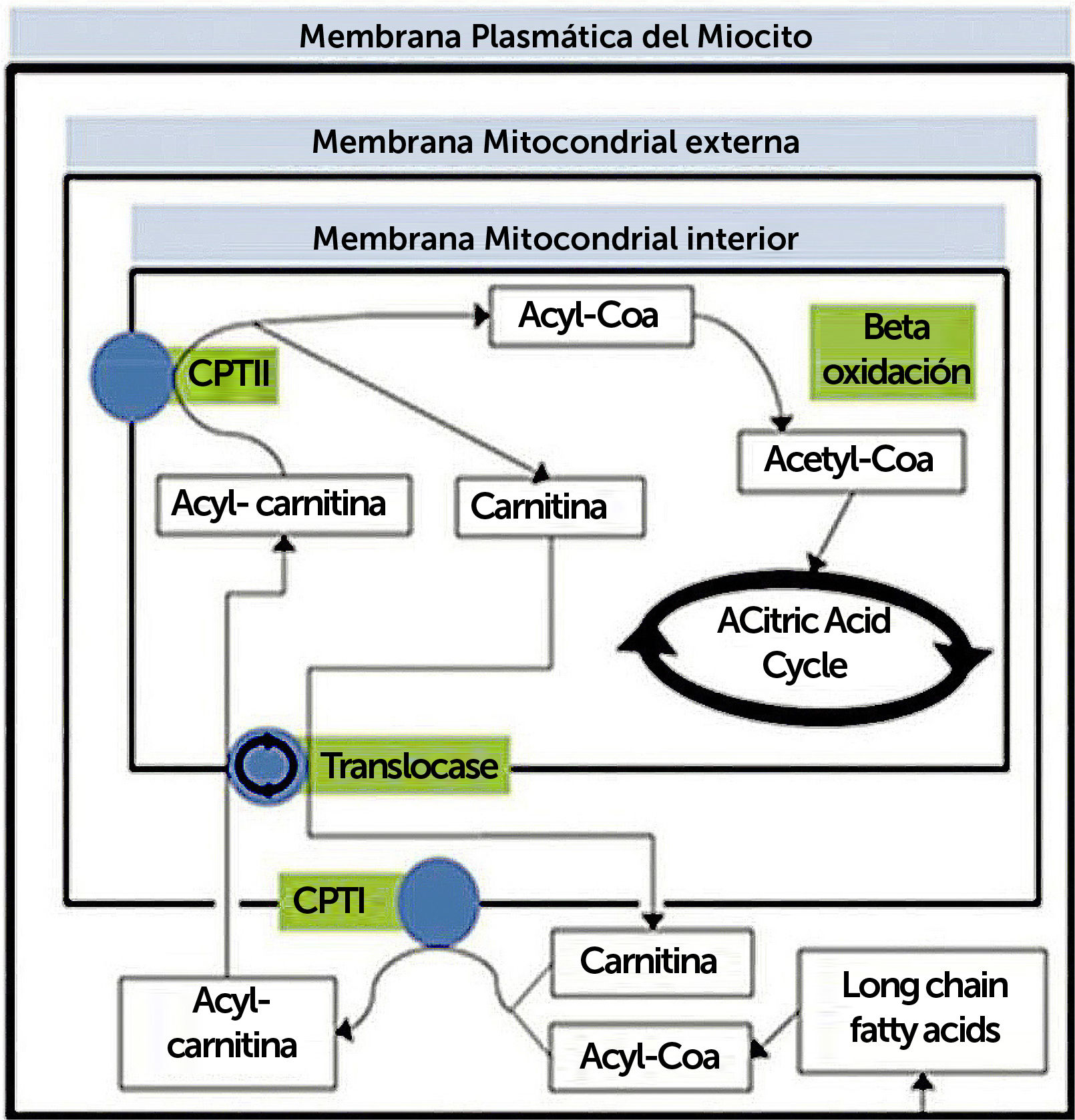
TRASTORNOS DEL METABOLISMO DE LOS LÍPIDOSProceso metabolico de trasporte y oxidación de ácidos grasos (Figura 2)
, ya que la membrana interna de la mitocondria es impermeable, por lo que deben unirse a una molécula de carnitina. Por otro lado, la beta-oxidación, en la matriz mitocondrial, consiste una serie de reacciones que finalmente se traducen en producción de una molécula de acetil-CoA, el que ingresa al ciclo de Krebs uniéndose a oxaloacetato para formar citrato. (Traducido de: Lilleker JB, Keh YS, Roncaroli F, et al. Pract Neurol 2018;18:14–265).")
Resumen de proceso metabólico de transporte y oxidación de ácidos grasos. CPT, carnitina palmitotransferasa
La oxidación de ácidos grasos ocurre en la mitocondria. Los ácidos grasos de cadena media y corta pueden entrar a la mitocondria directamente, pero no los de cadena larga (entre 14 y 20 carbonos), ya que la membrana interna de la mitocondria es impermeable, por lo que deben unirse a una molécula de carnitina. Por otro lado, la beta-oxidación, en la matriz mitocondrial, consiste una serie de reacciones que finalmente se traducen en producción de una molécula de acetil-CoA, el que ingresa al ciclo de Krebs uniéndose a oxaloacetato para formar citrato.
(Traducido de: Lilleker JB, Keh YS, Roncaroli F, et al. Pract Neurol 2018;18:14–265).
Pueden deberse a:
- -
Alteraciones en la degradación de triglicéridos en miocitos
- -
Alteraciones en la disponibilidad y transporte de carnitina
- -
Alteración en el transporte mitocondrial de ácidos grasos de cadena larga o en la β-oxidación de los ácidos grasos.
Las presentaciones clínicas abarcan un amplio espectro, con cuadros caracterizados por síntomas restringidos al músculo esquelético (algunos con debilidad permanente y otros con síntomas episódicos y rabdomiolisis) o síntomas multisistémicos (cardiomiopat-a progresiva, encefalopatía recurrente con hipoglicemia no cetósica, crisis epilépticas, discapacidad intelectual y muerte súbita entre otros).
Los principales cuadros con debilidad permanente y depósito de lípidos intracelulares en la tinción con O-Red-Oil o Sudan incluyen:
- -
Deficiencia primaria de Carnitina (Transportador de Carnitina)
- -
Enfermedad por depósito de lípidos neutros (NSLD)
- -
Deficiencia múltiple de Acil-CoA deshidrogenasa (MAD).
Los trastornos episódicos se asocian en general a lipidosis leve a moderada e implican la alteración de oxidación de ácidos grasos. Las causas principales son:
- -
-Deficiencia de carnitina palmitoil transferasa II (CPT II)
- -
-Deshidrogenasa de acil-CoA de cadena muy larga (VLCAD sigla en inglés)
- -
-Deficiencia trifuncional de proteínas mitocondriales y la de fosfatasa del ácido fosfatídico (Lipina1).
El estudio recomendado ante sospecha de trastorno del metabolismo lipídico se resume en la Tabla 422.
Estudio ante sospecha alteración metabolismo lipídico
| - Laboratorio básico: Electrolitos, transaminasas, nitrógeno ureico, creatinina, cetonuria |
| - Ácidos orgánicos en orina, incluyendo acilglicinas |
| - Lactato en sangre / líquido cefalorraquídeo (LCR) |
| - Carnitina total y libre en plasma |
| - Perfil acilcarnitinas en plasma (Espectrometría Masas en Tándem) |
| - CK total y MB sérica |
| - Cultivo fibroblasto para análisis enzimático |
| - Análisis genético molecular |
| - Biopsia muscular (Histología y análisis enzimático) |
De presentación en el adulto joven, es caracterizada por debilidad proximal lentamente progresiva, pero con síntomas de retraso motor y baja tolerancia al ejercicio en la infancia. Es causada por una mutación en el gen PNLPA2, de herencia autosómica recesiva, que codifica para adipo-triglicérido-lipasa (Desnutrina o ATGL), en el cromosoma 11p1523. La CK está elevada hasta 5 veces. La biopsia muscular muestra acumulación masiva de gotas lipídicas en fibras tipo 1 y 2, incluso en pacientes sin clínica de miopatía, y en leucocitos (anomalía de Jordan)24.
Trastornos del sistema de la carnitinaLa oxidación de ácidos grasos (AG) ocurre en la mitocondria (excepto los de cadena muy larga en peroxisomas), principalmente a través de oxidación. El primer paso es la activación a ácido graso-acil-CoA (AG-acil-CoA) en el retículo endoplásmico y la membrana externa mitocondrial, reacción catalizada por acil-CoA sintetasa. Los ácidos grasos de cadena media y corta pueden entrar a la mitocondria directamente, pero no los de cadena larga (entre 14 y 20 carbonos), ya que la membrana interna de la mitocondria es impermeable a estos AG-acil-CoA, por lo que deben unirse a una molécula de carnitina, proceso catalizado por la enzima carnitina palmitoil-transferasa I (CPT I), que elimina el coenzimo A de la molécula de acil-CoA y une el acil a la carnitina formando acilcarnitinas. El CoA queda libre para activar otro ácido graso, y la acilcarnitina es transferida a la matriz mitocondrial mediante una translocasa, situada en la membrana mitocondrial interna, y la carnitina palmitoil-transferasa II (CPT II) le une una molécula de CoA al ácido graso, regenerando el acil-CoA25.
Los ácidos grasos de más de 20 carbonos (de cadena muy larga) son oxidados en los peroxisomas, pero estos carecen de ciclo del ácido cítrico y cadena respiratoria, por lo que los productos de la β-oxidación en los peroxisomas, que incluyen acetil-CoA, propionil-CoA y otras acil-CoAs, también NADH, deben ser trasladados a las mitocondrias para la oxidación completa a CO2 y H2O y re-oxidación del NADH a NAD+26.
La L-carnitina es un compuesto hidrosoluble presente en casi todas las células humanas, sintetizada en hígado y riñón y almacenada en su mayoría en el músculo esquelético y cardíaco. La dieta (carnes rojas y lácteos) aporta alrededor del 75% de la carnitina necesaria.
Los trastornos del ciclo de la carnitina se pueden dividir en:
- -
Deficiencias primarias: deficiencia muscular, renal o sistémica de carnitina
- -
Deficiencias de carnitina secundarias a otro trastorno (hepático, renal, etc.).
- -
Deficiencia de CPT I y II
- -
Deficiencia de carnitina-acilcarnitina translocasa
a) Deficiencia primaria de carnitina (defecto del transportador de carnitina)
Es causada por un defecto en el transportador de carnitina, OCTN2, en la membrana plasmática de varios tejidos, incluyendo músculo, corazón y riñón, pero no del hígado. La carnitina es necesaria para la translocación de ácidos grasos en la membrana mitocondrial, por lo que se produce una disminución de oxidación de ácidos grasos y aumento en la formación de triglicéridos con depósito de lípidos en miocitos27.
De herencia autosómica recesiva, tiene dos formas de presentación importantes de reconocer, ya que el tratamiento es efectivo.
Forma miopática
Se caracteriza por hipotonía, debilidad proximal y miocardiopatía, con una progresión usualmente lenta de inicio variable entre el primer año y la segunda década de vida.
Generalmente existe aumento de niveles séricos de CK y la biopsia muscular muestra acúmulo de lípidos especialmente en fibras de tipo I y bajos niveles de carnitina en el análisis bioquímico28. Los niveles de carnitina plasmática son normales.
Forma sistémica
También presenta compromiso muscular con depósito de lípidos en fibras musculares, pero con afectación de otros sistemas. Se manifiesta en la infancia con fatigabilidad, vómitos, dolor abdominal, talla baja y deficiencia de peso. Cursa con episodios de hipoglicemia no cetósica, encefalopatía y hepatomegalia29. La carnitina plasmática total y libre se encuentra en niveles de menos del 10% de los controles, pero no se detectan acilcarnitinas aumentadas, y la carnitina total en el músculo se reduce a menos del 5% de los controles30. Se han reportado mutaciones en el gen SCL22A5 que codifica para el transportador OCTN231.
El tratamiento en ambas formas puede revertir los síntomas, y consiste en la administración de una dieta pobre en grasa (especialmente ácidos grasos de cadena larga) y suplemento de L-carnitina 2 a 4 grs/día (niños: 100 mg/kg/día)32. En los casos de descompensación se debe usar glucosa ev.
b) Deficiencia de Carnitina-Palmitoil-Transferasa II (CPT II) La deficiencia de actividad de la CPT II es el trastorno hereditario del metabolismo lipídico más común que afecta el músculo esquelético, con una prevalencia de 1-9/10000034.
La forma miopática clásica se caracteriza clínicamente por episodios recurrentes de mialgias y/o debilidad muscular y rabdomiolisis precipitados por ejercicio prolongado, ayuno, dieta pobre en hidratos de carbono y alta en grasa, fiebre e infecciones. Entre crisis los pacientes presentan una fuerza normal. Aunque en ocasiones es considerada como de presentación más frecuente en el adulto, habitualmente los pacientes presentan antecedentes de mialgias o mioglobinuria desde la infancia alcanzando hasta un 60% de los casos un inicio en la infancia.
Otros fenotipos menos frecuentes son la forma neonatal letal con grave compromiso hepático y muscular e hipoglicemia hipocetósica, y una forma infantil, que cursa con episodios de insuficiencia hepática aguda con hepatomegalia transitoria, hipoglicemia hipocetósica, coma y convulsiones.
El gen es CPT2, y respecto a la correlación genotipo-fenotipo, en aproximadamente el 70% de pacientes con la forma miopática existe una mutación sin sentido en p.Ser113Leu33 y aquellas variantes patogénicas en este gen que resultan en una proteina trunca o degradación de RNM mensajero, están asociadas con las formas más severas de la enfermedad34.
Durante el episodio agudo se detecta aumento de CK, la que vuelve a los valores normales después de la crisis. La biopsia muscular no muestra alteraciones morfológicas, pudiendo ser normal hasta en 50% de los casos. El examen de mayor utilidad es el estudio de acilcarnitinas mediante espectrometría de masas en tándem, que muestra un perfil característico con elevación principalmente de compuestos de 16 a 18 carbonos; sin embargo, un estudio normal no descarta completamente el diagnóstico, sobretodo si el paciente está en una fase asintomática35,36.
El diagnóstico confirmatorio actual incluye el estudio genético del gen CPT2 ante la sospecha clínica y de laboratorio.
El tratamiento se basa en la indicación de comidas frecuentes, dieta con restricción de triglicéridos de cadena larga, suplementada con triglicéridos se cadena media e hidratos de carbono, incluyendo aporte de almidón crudo. Evitar el ejercicio prolongado o en condiciones de ayuno o infecciones intercurrentes. Se recomienda la infusión de glucosa previo y durante la anestesia general.
Defectos intramitocondriales de la β-oxidaciónLa β-oxidación en la matriz mitocondrial consiste en cuatro reacciones que finalmente se traducen en producción de una molécula de acetil-CoA por cada par de carbonos restados al grupo acilo, que ingresa al ciclo de Krebs uniéndose a oxaloacetato para formar citrato.
Las cuatro principales enzimas implicadas en la β-oxidación son:
- -
Acetil-CoA deshidrogenasa
- -
Enoil-CoA hidratasa
- -
Hidroxi acetil-CoA deshidrogenasa
- -
Ketoacil-CoA tiolasa
Los cuadros de deficiencia de deshidrogenasas de acil-CoA de cadena muy larga (VLCAD), larga (LCAD), mediana (MCAD) y corta (SCAD) pueden presentarse con síntomas sistémicos de gravedad variable, como episodios de hipoglicemia, disfunción hepática, letargia, miocardiopatía, o un compromiso más restringido a signología muscular. La CK en reposo es normal. El diagnóstico precoz mediante tamizaje neonatal o ante la presencia de clínica sugerente es fundamental para reducir morbimortalidad asociada a estos cuadros, evitando principalmente el ayuno.
Deficiencia de fosfatasa de ácido fosfatídico (LIPIN1)La fosfatasa del ácido fosfatídico convierte el ácido fosfatidico en diacilglicerol en la vía del triacilglicerol. Existen 3 isoformas: LIPIN1 se expresa en musculo y tejido adiposo, LIPIN 2 en tejido adiposo (lo que explica la ausencia de lipodistrofia) y LIPIN 3. La mutación en LIPIN1, autosómica recesiva (locus 2p25.1) produce un defecto en la composición de fosfolípidos en la membrana mitocondrial interna y acumulación de lisofosfolípidos, que actúan como detergentes, lo que favorece la degradación muscular.
Clínicamente se manifiesta por episodios de mioglobinuria recurrente en niños entre los 15 meses y los 7 años de edad, generalmente precipitado por enfermedades febriles, ayuno, ejercicio intenso o condiciones de estrés catabólico, cursando con CK muy elevadas (habitualmente sobre 10000 U/L); se describe que un tercio de los pacientes mueren en los episodios de rabdomiólisis. El uso de sobre hidratación y altas cargas de glucosa ha permitido disminuir la duración de episodios desde 10 a 5 días en promedio35.
Entre los episodios los pacientes tienen examen neurológico y CK normales o levemente elevadas (hasta 700 U/L)37,38.
DEFECTOS DE CADENA RESPIRATORIA MITOCONDRIALLas enfermedades mitocondriales se refieren en sentido estricto a los trastornos primarios de la fosforilación oxidativa (OXPHOS), es decir, por deficiencia objetiva en elementos de la cadena respiratoria o mutación patogénica del ADN mitocondrial (ADNmit).
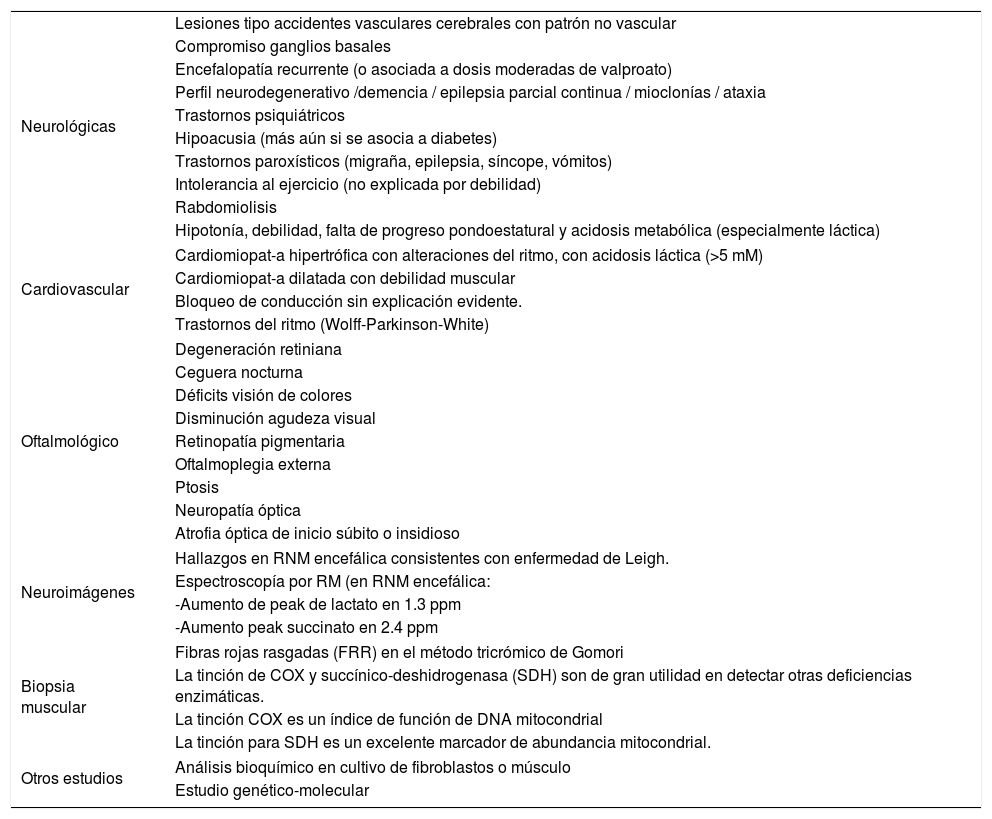
La sospecha clínica debe plantearse en todo paciente con asociación inexplicada de signos y síntomas y/o compromiso de más de 2 o 3 sistemas, siendo algunas presentaciones de mayor especificidad (Tabla 5)39.
Elementos clínico-diagnósticos asociados a Enfermedades Mitocondriales
| Neurológicas | Lesiones tipo accidentes vasculares cerebrales con patrón no vascular |
| Compromiso ganglios basales | |
| Encefalopatía recurrente (o asociada a dosis moderadas de valproato) | |
| Perfil neurodegenerativo /demencia / epilepsia parcial continua / mioclonías / ataxia | |
| Trastornos psiquiátricos | |
| Hipoacusia (más aún si se asocia a diabetes) | |
| Trastornos paroxísticos (migraña, epilepsia, síncope, vómitos) | |
| Intolerancia al ejercicio (no explicada por debilidad) | |
| Rabdomiolisis | |
| Hipotonía, debilidad, falta de progreso pondoestatural y acidosis metabólica (especialmente láctica) | |
| Cardiovascular | Cardiomiopat-a hipertrófica con alteraciones del ritmo, con acidosis láctica (>5 mM) |
| Cardiomiopat-a dilatada con debilidad muscular | |
| Bloqueo de conducción sin explicación evidente. | |
| Trastornos del ritmo (Wolff-Parkinson-White) | |
| Oftalmológico | Degeneración retiniana |
| Ceguera nocturna | |
| Déficits visión de colores | |
| Disminución agudeza visual | |
| Retinopatía pigmentaria | |
| Oftalmoplegia externa | |
| Ptosis | |
| Neuropatía óptica | |
| Atrofia óptica de inicio súbito o insidioso | |
| Neuroimágenes | Hallazgos en RNM encefálica consistentes con enfermedad de Leigh. |
| Espectroscopía por RM (en RNM encefálica: | |
| -Aumento de peak de lactato en 1.3 ppm | |
| -Aumento peak succinato en 2.4 ppm | |
| Biopsia muscular | Fibras rojas rasgadas (FRR) en el método tricrómico de Gomori |
| La tinción de COX y succínico-deshidrogenasa (SDH) son de gran utilidad en detectar otras deficiencias enzimáticas. | |
| La tinción COX es un índice de función de DNA mitocondrial | |
| La tinción para SDH es un excelente marcador de abundancia mitocondrial. | |
| Otros estudios | Análisis bioquímico en cultivo de fibroblastos o músculo |
| Estudio genético-molecular | |
RNM: resonancia nuclear magnética.
Las formas más frecuentes de presentación de las enfermedades mitocondriales con compromiso muscular predominante son la deficiencia de citocromo C-Oxidasa (COX), la oftalmoplegia externa crónica progresiva (CPEO), síndrome de Kearns Sayre, deficiencia de coenzima Q10 (CoQ10) y un cuadro asociado a deleciones múltiples del ADN mit.
Como otras miopatías metabólicas, las miopatías mitocondriales pueden presentar síntomas episódicos, síntomas estáticos o la combinación de ambos, con mialgias con ejercicio con o sin mioglobinuria, hasta síntomas graves como disnea asociada a ejercicio o rabdomiolisis.
La debilidad puede ser de inicio en los primeros años de vida tienen una presentación clínica grave, como la deficiencia de COX, caracterizada por debilidad e hipotonía severas, con insuficiencia respiratoria en el periodo de recién nacido y un curso habitualmente fatal dentro del primer año de vida.
En el caso del déficit primerio de coenzima Q10, se describe la triada clásica de:
- 1)
intolerancia al ejercicio y mioglobinuria recurrente;
- 2)
disfunción del sistema nervioso central, con convulsiones o discapacidad cognitiva y
- 3)
biopsia muscular con fibras rojas rasgadas (FRR) y aumento marcado de gotas de lípidos40.
En el síndrome de Kearns Sayre la debilidad es generalizada, asociada a oftalmoplegia externa crónica progresiva (CPEO), retinopatía pigmentaria, defectos de conducción cardiaca (que puede llegar al bloqueo completo requiriendo marcapasos), ataxia y aumento de proteínas en el LCR. Se manifiesta en la primera a segunda década de la vida, con talla baja, fatigabilidad y episodios de encefalopatía. Pueden presentar deficiencia de hormona de crecimiento, hipoparatiroidismo y/o diabetes mellitus. Es habitual la acidosis láctica (en sangre y LCR). En los estudios de neuroimágenes destaca compromiso de sustancia blanca y ganglios basales. La biopsia muscular puede mostrar FRR.
En el caso de deleciones múltiples o depleción del ADN mit, se describen mutaciones en timidina quinasa 2 (TK2) que se reporta como causa de un síndrome miopático temprano y fatal, por depleción del ADN mit. Insuficiencia respiratoria con o sin encefalopatía, miopatía crónica progresiva en el adulto u oftalmoplegia externa se han asociado a deleciones múltiples del ADN mit39. La depleción severa del ADN mit, de genética heterogénea, se considera una de las formas más comunes en la infancia de las deficiencias de la cadena respiratoria.
Tratamiento
Con la excepción de algunos pacientes con deficiencia primaria de CoQ10 que muestran una mejoría dramática después de la suplementación con CoQ10 y riboflavina41, en otros cuadros los efectos reportados son en general modestos y subjetivos. Se ha planteado que idebenona y parabenzoquinona, análogos sintéticos de CoQ10, podrían tener mejores resultados42. La administración de coenzima Q10, creatina, acido lipoico y tiamina se deben incluir en el síndrome de Kearns Sayre43, asociado a la instalación de marcapasos, dado que defectos de conducción cardíaca son la causa de muerte más frecuente44.
TRASTORNOS DEL METABOLISMO DE LAS PURINASDéficit de mioadenilato deaminasa (DMA)Se manifiesta por mialgias post-ejercicio, calambres, e intolerancia al ejercicio, de inicio en la infancia, adolescencia o edad adulta. Evoluciona con síntomas moderados a severos durante los primeros años, pero luego el curso clínico generalmente se estabiliza. Es un trastorno hereditario autosómico recesivo producido por mutaciones en el gen AMPD1, que afectan el ciclo de nucleótidos de purinas. La mayoría de los pacientes son homocigotos y la mutación más frecuente en este gen es la C34T, presente en cerca del 1% de la población caucásica, pero la mayoría asintomática. El diagnóstico se basa en histoquímica y análisis bioquímico en músculo, o en la identificación de las mutaciones causales. Si la curva de amonio es plana, respecto al ascenso de lactato post-ejercicio en el test de ejercicio isquémico, se plantea este trastorno.
Se ha descrito déficit de la actividad enzimática de la isoforma de eritrocitos de AMPdeaminasa en pacientes con bajos niveles de ácido úrico, sin signos clínicos evidentes. Junto con aquello, el DMA se a asociado con otros trastornos neuromusculares, déficit que en este caso podría ser precipitado por cambios patológicos o en sinergismo con otros trastronos metabólicos involucrados.
Se ha reportado mejoría de síntomas tras ingesta de D-ribosa, con efectos transitorios45.
ENFOQUE CLÍNICO DE LAS MIOPATÍAS METABÓLICASLa forma de presentación puede sugerir el diagnóstico (Figura 3). Los síntomas que aparecen durante el ejercicio inicial sugieren un defecto del metabolismo de hidratos de carbono, pero si se presentan tras el ejercicio prolongado son más indicativos de un trastorno del metabolismo de los lípidos.

Dependiendo de la etiología, pueden presentarse con signología agregada a los síntomas musculares, como encefalopatía (compromiso de conciencia), crisis epilépticas, mioclonías o ataxia. En los adultos, el motivo de consulta generalmente se refiere a fatiga muscular, dolor, calambres o rigidez muscular. En los niños pequeños se pueden presentar como retraso del desarrollo, hipotonía o anormalidad en la adquisición de hitos motores. La mayoría de los trastornos primarios del metabolismo son hereditarios, por lo que se debe hace una cuidadosa anamnesis familiar.
Al examen físico puede encontrarse grados variables de debilidad, habitualmente de predominio proximal. La presencia de oftalmoparesia o ptosis debe hacer sospechar una miopatía mitocondrial. La rigidez o contracción muscular mantenida post-ejercicio es característica de las glicogenosis.
Diagnóstico
Las miopatías metabólicas constituyen cuadros de difícil confirmación, por lo que se requiere un enfrentamiento ordenado en relación con los procedimientos diagnósticos necesarios.
Laboratorio
La medición de CK sérica se considera uno de los exámenes iniciales más útiles en la evaluación de las miopatías metabólicas, especialmente en el adulto, en que la consulta por fatiga y mialgias es muy frecuente. Otros exámenes deben realizarse de acuerdo con la historia y examen físico, pero es útil realizar un panel básico inicial ante la sospecha de alguno de estos cuadros (Tabla 6).
Exámenes básicos a realizar frente a sospecha de miopatía metabólica
| Creatinkinasa total y fracción MB |
| Transaminasas |
| Electrolitos plasmáticos |
| Hormonas tiroideas |
| Velocidad de sedimentación |
| Lactacidemia basal y post-ejercicio o post-sobrecarga de glucosa |
| Amonio |
| Mioglobinuria |
| Perfil de Acilcarnitinas (Espectrometría de masas en Tándem) |
| Carnitina libre y esterificada |
Si la historia y/o examen sugieren un trastorno del metabolismo de hidratos de carbono, el test de ejercicio isquémico puede ser de utilidad en aclarar el diagnóstico, que se confirmará mediante análisis enzimáticos en sangre, músculo o fibroblastos y/o análisis genético molecular.
Es necesario buscar dirigidamente compromiso de otros sistemas: evaluación cardiológica, renal, oftalmológica, exámenes de función hepática, función respiratoria. Estos estudios no solo pueden aportar al diagnóstico, sino que son fundamentales para el tratamiento y detección de complicaciones.
Electrofisiología
Los estudios de conducción nerviosa son en general normales en las miopatías metabólicas. La electromiografía es útil en las miopatías metabólicas que cursan con compromiso muscular persistente, donde pueden observarse potenciales miopáticos. En la enfermedad de McArdle durante el episodio de mialgias puede demostrarse silencio eléctrico que permite certificar el estado de contractura muscular.
Biopsia muscular
La biopsia muscular puede brindar información de gran utilidad, pero en muchos casos no es necesaria ya que existen pruebas bioquímicas o genético-moleculares más específicas y accesibles. Si está indicada, debe tomarse la precaución de realizar un manejo adecuado del tejido y contar con las técnicas histoquímicas o bioquímicas necesarias para el análisis.
Estudio genético moleculares
Considerando los costos actuales de estos estudios, y la certeza diagnóstica, es que los estudios genéticos moleculares han ido reemplazando otras evaluaciones. En caso de tener sospecha clínica elevada de un tipo de miopatía metabólica, se puede solicitar el estudio molecular específico directamente, ya que entrega mayor certeza y permite realizar un asesoramiento genético si esto fuera necesario.
Se debe tener en consideración que están disponibles distintos “paneles genéticos” que estudian un grupo de genes de acuerdo a la manifestación clínica/síntoma principal, ej. panel genético de rabdomiolisis, cuyo costo es más bajo que estudios de estudios genéticos más amplios como Whole Genome Sequencing (WGS) o Whole Exome Sequencing (WES).
CONCLUSIONESEl diagnóstico diferencial de las miopatías metabólicas es amplio, por lo que debe ser enfocado de acuerdo a las características del cuadro clínico, basadas en una detallada anamnesis y examen físico, acompañado de un estudio sistémico adecuado. El estudio de laboratorio básico en estas patologías es un estudio disponible en la mayoría de los centros clínicos del país, que permite orientar un estudio de segunda línea que sea del mayor costo/efectividad y certeza posible. La importancia diagnóstica de estas patologías radica en un potencial tratamiento que alivie los síntomas y que evite complicaciones a largo plazo, además de en un adecuado consejo genético.
Declaración de interésNo hay conflicto de interes que declarar por los autores: no se recibió financiamiento por escritura de artículo.