




Las distrofias musculares son un grupo de trastornos hereditarios, degenerativos, progresivos del músculo estriado, cuya manifestación cardinal es la debilidad de la musculatura estriada esquelética. En esta revisión se describirá conceptualmente la clasificación, la patogénesis, las manifestaciones clínicas, el diagnóstico diferencial y el enfoque terapéutico de las distrofias musculares más comunes que afectan al paciente adulto.
The muscular dystrophies are a group of inherited, progressive, degenerative muscular disorders whose cardinal manifestation is the weakness of skeletal muscles. In this review we will conceptually describe its classification, the pathogenesis, the most relevant clinical features, its differential diagnosis and the therapeutic approach of the most common muscular dystrophies in the adult patient.
Las distrofias musculares son un grupo heterogéneo de enfermedades del músculo estriado causadas por mutaciones genes que determinan la reducción, ausencia o disfunción de proteínas esenciales para la estabilidad estructural y funcional de las fibras musculares esqueléticas, lo que conduce a la destrucción y debilidad muscular de forma progresiva1. Clínicamente, las distrofias musculares se caracterizan por una debilidad muscular progresiva de las extremidades, el tronco y la cara en proporciones y severidad variables, pudiendo involucrar, en algunas formas específicas, la musculatura respiratoria, cardíaca y los músculos craneofaciales (oculomotores, deglución, masticatorios). En algunos casos, la afección muscular es parte de un síndrome multisistémico, como es el caso de la distrofia miotónica1,2. La severidad, edad de comienzo, la evolución, así como las complicaciones y el pronóstico de las distrofias son muy variables dependiendo del gen mutado y/o de la mutación. A nivel histopatológico, una distrofia muscular se define por una combinación de necrosis y regeneración, asociado a aumento del tejido conectivo intersticial, los que pueden variar según el momento evolutivo del cuadro. Dichas características permiten diferenciar a las distrofias musculares de otras miopatías no distróficas, como las miopatías congénitas, metabólicas, inflamatorias, entre otras. Este fenómeno de destrucción muscular lleva con el tiempo, a un reemplazo del músculo por tejido fibroadiposo1,2.
A partir de la identificación del gen causal de la distrofinopatía (distrofia muscular de Duchenne/Becker) en 19873, ha habido un incremento exponencial en la identificación de mutaciones causales de distintas formas de distrofia muscular. Entre las distrofias musculares más comunes que se presentan en la edad adulta se encuentran las distrofinopatías, como la distrofia muscular de Becker (DMB)4, la distrofia miotónica tipos 1 y 2 (DM1, DM2)2, la distrofia facioescápulohumeral (FSHD)5, la distrofia de Emery–Dreifuss (EDMD)6,7, la distrofia muscular oculofaríngea (OPMD)8; así como un grupo de unas 30 distrofias musculares que afectan predominantemente las cinturas pélvica y escapular, y que han sido agrupadas bajo la denominación de distrofia muscular de cinturas o LGMD9, por su sigla en inglés (limb-girdle muscular dystrophies).
Los adultos pueden padecer una distrofia muscular que puede haber comenzado en etapas más tempranas de la vida, como también una distrofia puede comenzar a manifestarse en la adultez. Dado la variabilidad en la edad de presentación así como de las manifestaciones clínicas de las distrofias musculares, es difícil hacer una revisión que solamente abarque las distrofias que se manifiestan en los adultos, sin aludir o mencionar algunas formas de distrofia que se presentan en pacientes pediátricos o adolescentes. Igualmente la distinción entre distrofias y miopatías no distróficas no es siempre precisa, y hay una inevitable sobreposición de conceptos. En este sentido, trataremos de enfocarnos en las miopatías distróficas más comunes que se observan en el adulto, conscientes de que la definición de las distrofias musculares como grupo es imprecisa.
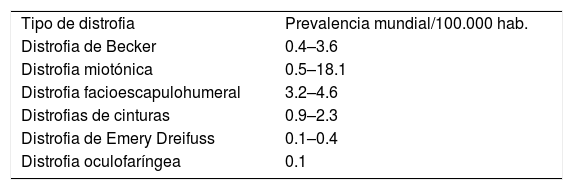
EPIDEMIOLOGÍATodas las distrofias musculares caen dentro de la categoría de enfermedades raras por su baja prevalencia, la que a nivel mundial se ha estimado entre 19.8–25.1/100000 tomando las distrofias musculares como grupo. Sin embargo, la mayoría de los estudios provienen de países desarrollados y es difícil hacer extrapolaciones precisas10. En Chile no hay estadísticas fidedignas del número de pacientes afectados por distrofia muscular, se estima que unos 5000 pacientes estarían afectados por miopatías hereditarias, pero es posible que las miopatías en general estén subdiagnosticadas11. Las prevalencias descritas por cada 100000 habitantes de las distrofias musculares más frecuentes en el adulto se detallan en la Tabla 110. Una distrofia muscular puede ser rara en una región del planeta, pero puede no serlo en otra. La prevalencia relativa de las distintas formas de distrofia muscular varía en distintas regiones geográficas y según el grupo étnico en estudio. En Chile se han comunicado casos y pequeñas series de pacientes con las distrofias musculares más prevalentes13–17. En los últimos años, a través de una pesquisa más sistematizada, se ha identificado un número creciente de pacientes con disferlinopatía (Miopatía de Miyoshi/LGMD2B) y calpainopatías (LGMD2A), y hay evidencia preliminar de un posible efecto fundador de mutaciones en estos genes en Chile18,19.
Prevalencia relativa estimada de los distintos subtipos de distrofias musculares por 100.000 habitantes
| Tipo de distrofia | Prevalencia mundial/100.000 hab. |
| Distrofia de Becker | 0.4–3.6 |
| Distrofia miotónica | 0.5–18.1 |
| Distrofia facioescapulohumeral | 3.2–4.6 |
| Distrofias de cinturas | 0.9–2.3 |
| Distrofia de Emery Dreifuss | 0.1–0.4 |
| Distrofia oculofaríngea | 0.1 |
Tomado de estudios de prevalencia para la población general10.
La patogenia de las distrofias musculares es diversa y compleja, y sobrepasa el objetivo de esta revisión. De forma conceptual, las distrofias musculares son causadas más comúnmente por la ausencia, reducción o disfunción de proteínas esenciales para la estabilidad estructural y funcional de las fibras musculares esqueléticas, lo que conduce a la destrucción y debilidad muscular de forma progresiva21. La fisiopatología de cada distrofia muscular es diferente dependiendo de la proteína afectada, y a veces incluso del tipo de mutación que la determina aun dentro de un mismo gen. Las proteínas mutadas pueden ser parte estructural y/o funcional de cualquier componente de la fibra muscular (sarcolema, sarcómero, núcleo, entre otros.) así como de la matriz extracelular que las rodea1,20,21. Algunas de estas proteínas, como la distrofina y su complejo de glicoproteínas asociadas (sarcoglicanos, distroglicanos, distrobrevina, sintrofina, entre otras), tienen una función de anclaje del aparato contráctil del citoesqueleto a la matriz extracelular; otras, como por ejemplo la emerina y la lamina A/C estabilizan la membrana nuclear1,6,20,21. Las mutaciones de proteínas sarcoméricas, en particular las que tienen relación con la línea Z, producen desorganización del sarcómero, provocando una agregación de las proteínas que se acumulan dentro de la fibra impidiendo su funcionamiento normal. En otras distrofias, las mutaciones afectan a enzimas que participan en la glicosilación de proteínas estructurales, como es el caso de los distroglicanos, y que al no estar glicosilados son eliminados por la célula, provocando su deficiencia. También pueden estar afectadas proteínas involucradas en diversos procesos (calpaína, disferlina, etc.). Algunas mutaciones de proteínas de la matriz extracelular como el colágeno VI (miopatía de Ullrich/Bethlem) y laminina (distrofia congénita) también pueden causar una distrofia muscular1,22. Asimismo, en algunas distrofias musculares como la distrofia miotónica y la distrofia oculofaríngea, la expansión de residuos nucleotídicos (tripletes, cuatripletes) se acumulan en las células en niveles tóxicos, determinando un malfuncionamiento de la transcripción y traducción génicas, provocando diferentes alteraciones a nivel muscular, del SNC, así como en otros tejidos y órganos.
CARACTERÍSTICAS CLÍNICASLos síntomas y signos comunes a todas las distrofias derivan de la disfunción del músculo estriado y la distribución de los músculos involucrados. Interesantemente, en las distrofias musculares no se afectan todos los músculos del cuerpo de forma uniforme, esto hace que haya una distribución particular de la afección muscular en cada tipo de distrofia, en particular en etapas iniciales de la enfermedad1.
Los hallazgos comunes son la debilidad y atrofia muscular progresiva e indolora de grupos musculares determinados, que progresa a otros grupos musculares específicos o puede generalizarse1 . Muchas veces, pese a la debilidad evidente, el paciente no se queja de la misma, ya que los síntomas se establecen lentamente, permitiendo una compensación muy eficiente de los déficits. En algunos casos esta hipofunción muscular se manifiesta inicialmente como intolerancia al ejercicio o fatigabilidad; además de la debilidad, otros síntomas musculares incluyen calambres, miotonía y mialgias. En otros casos, la consulta del paciente puede ser una manifestación indirecta de la debilidad, por dificultad respiratoria o infecciones respiratorias intercurrentes, disfagia, malformaciones esqueléticas, dolores articulares, así como síntomas cardíacos.
Si la debilidad predomina o comienza en la cintura pélvica y las extremidades inferiores, los pacientes suelen quejarse de caídas frecuentes, dificultad para subir escaleras, levantarse de una silla, correr, etc. La marcha de Trendelemburg o marcha miopática (anserina o de pato) muy común en las distrofias musculares, se produce por debilidad de los estabilizadores de la pelvis (glúteos medios). Cuando la debilidad predomina en la cintura escapular, hay dificultad para elevar los brazos sobre los hombros, peinarse, maquillarse, cargar objetos, etc., manifestaciones que pueden acompañarse de dificultades para cambiar de posición en la cama cuando se afectan los músculos axiales. En ocasiones, las manifestaciones están limitadas a un grupo muscular específico; por ejemplo, la musculatura cervical que se manifiesta como dificultad para mantener la cabeza erguida (síndrome de la cabeza caída o dropped head syndrome)22, o para flexionarla en decúbito. En la debilidad progresiva de la musculatura erectora dorsal hay una anteflexión del tronco, que puede llegar a ser permanente (camptocormia). La debilidad abdominal predispone a una hiperlordosis lumbar y puede producir un abdomen de aspecto prominente. Los familiares pueden indicar que el paciente duerme con los ojos abiertos, o que gesticula menos que el resto por debilidad facial (ausencia de sonrisa), como en la FSHD5.
La inspección de la cintura escapular es muy relevante para evidenciar un despegamiento escapular (escápula alada), muy evidente en la FSHD, donde característicamente es asimétrica; pero también puede verse en otras distrofias, como en la calpainopatía o las sarcoglicanoptías (LGMD2A, 2C-F)1,5,9. El acortamiento del músculo determina retracciones que adquieren formas características6,7. Los pacientes presentan los codos en semiflexión permanente, son incapaces de ponerse en posición de rezo o mantener el cuello hiperextendido con una rectificación del mismo16,22. El acortamiento del compartimiento posterior de la pierna hace que los pacientes caminen en puntas de pies; la retracción de los músculos paraespinales con pérdida de las curvaturas normales de la columna que impide la flexión del tronco, determinan una espina rígida1,22. Las alteraciones cardíacas incluyen trastornos de la conducción, miocardiopatía e insuficiencia cardíaca. La afección cardíaca es frecuente en algunas distrofias como en el caso de la distrofia miotónica (DM1), distrofinopatías y la distrofia de Emery Dreifuss1,2,4,6,7. En las distrofias musculares del adulto, el compromiso respiratorio es generalmente una manifestación tardía de la enfermedad. Este puede manifestarse como disnea de esfuerzo, ortopnea y cefalea matutina, y en algunas miopatías constituye el síntoma de presentación, como por ejemplo enfermedad de Pompe de comienzo tardío22,23.
Dependiendo de la sospecha diagnóstica, se debe explorar la presencia de cataratas, alteraciones del músculo liso intestinal y endocrinopatías (hipogonadismo, diabetes), todas presentes en la DM12. La piel puede verse afectada dando lugar a queloides y acantosis nigricans, como en las alteraciones del colágeno VI (miopatía de Bethlem)25,26.
A riesgo de sobresimplificar la gran diversidad clínica de las distrofias musculares y otras miopatías relacionadas, se pueden describir 6 patrones de presentación comunes, de acuerdo a la distribución predominante de los signos musculares1:
- 1.
Debilidad en cinturas escapular y pélvica. Es el principal y más frecuente. Característico de las distrofinopatías y las distrofias de cinturas (LGMD, DMB, etc.)1,9.
- 2.
Debilidad escápulo-peroneal: Lo característico es escápula alada y debilidad de la dorsiflexión del pie. Si es asimétrico e incluye paresia facial, es muy sugerente de FSHD. En cambio si se asocia a retracciones y es relativamente simétrico sugiere un síndrome de Emery Dreifuss (emerina o lamina A/C)5–7.
- 3.
Debilidad distal: Afecta la musculatura flexora o extensora del carpo y de los dedos en la extremidad superior, e intrínseca de la mano (distrofia miotónica, titinopatía) o en la musculatura distal de la extremidad inferior tríceps sural, dorsiflexores del pie, como en la miopatía distal de Miyoshi (disferlinopatía), donde hay atrofia del compartimiento posterior de las piernas y dificultad para caminar en puntas de pie15,27. Este patrón puede verse también en las miopatías miofibrilares27.
- 4.
Oculofaríngeo: Se observa ptosis y disfagia como síntomas más importantes y puede haber debilidad proximal. Típicamente no hay diplopía. Es característico de la distrofia oculofaríngea, también se ve en la distrofia miotónica, donde se acompaña de compromiso facial evidente, entre otras manifestaciones. El diagnóstico diferencial a considerar es la miastenia gravis y las miopatías mitocondriales8,17.
- 5.
Compromiso respiratorio temprano: Aunque no es muy común en las distrofias, incluimos este patrón ya que es importante reconocerlo porque es potencialmente tratable. Estos pacientes consultan por fatiga, disnea de esfuerzo, ortopnea y cefalea matutina, consultan múltiples especialistas antes de llegar al neurólogo, habitualmente con una espirometría con patrón restrictivo. Caen en este patrón, la enfermedad de Pompe de comienzo tardío (LOPD), la miopatía nemalínica, las miopatías miofibrilares y en algunos pacientes con distrofia miotónica tipo 123,27. En el diagnóstico diferencial deben considerarse la esclerosis lateral amiotrófica, la miastenia gravis y algunas formas de miopatía inflamatoria.
- 6.
Compromiso axial: Se manifiesta como cabeza caída/camptocormia que se puede observar en una miopatía esporádica de los erectores de la columna y como parte de algunas distrofias. En cambio la espina rígida se ve en las mutaciones de la selenoproteína 1, emerina y lamina (LGMD1B) entre otras miopatías6,7,22.
De forma habitual, los pacientes no presentan alteraciones sensitivas, cerebelosas o neurovegetativas, o si estas están presentes no son la principal afección, la presencia de estas en un paciente con sospecha de distrofia muscular, debe hacer dudar del diagnóstico de un trastorno muscular primario. Los reflejos osteotendíneos suelen estar presentes, son normales o pueden estar débiles o abolidos si el músculo efector está muy atrófico, lo que dependerá de la distribución y la gravedad de la afección (Tabla 2).
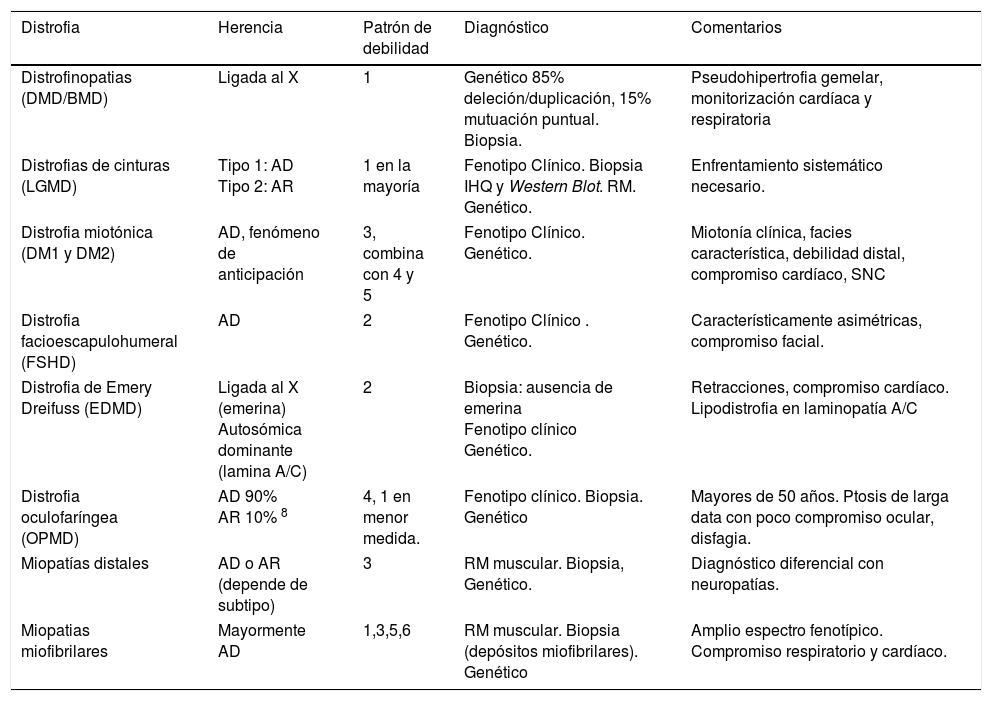
Sinopsis de las principales características de las distrofias musculares más frecuentes en el adulto
| Distrofia | Herencia | Patrón de debilidad | Diagnóstico | Comentarios |
|---|---|---|---|---|
| Distrofinopatias (DMD/BMD) | Ligada al X | 1 | Genético 85% deleción/duplicación, 15% mutuación puntual. Biopsia. | Pseudohipertrofia gemelar, monitorización cardíaca y respiratoria |
| Distrofias de cinturas (LGMD) | Tipo 1: AD Tipo 2: AR | 1 en la mayoría | Fenotipo Clínico. Biopsia IHQ y Western Blot. RM. Genético. | Enfrentamiento sistemático necesario. |
| Distrofia miotónica (DM1 y DM2) | AD, fenómeno de anticipación | 3, combina con 4 y 5 | Fenotipo Clínico. Genético. | Miotonía clínica, facies característica, debilidad distal, compromiso cardíaco, SNC |
| Distrofia facioescapulohumeral (FSHD) | AD | 2 | Fenotipo Clínico . Genético. | Característicamente asimétricas, compromiso facial. |
| Distrofia de Emery Dreifuss (EDMD) | Ligada al X (emerina) Autosómica dominante (lamina A/C) | 2 | Biopsia: ausencia de emerina Fenotipo clínico Genético. | Retracciones, compromiso cardíaco. Lipodistrofia en laminopatía A/C |
| Distrofia oculofaríngea (OPMD) | AD 90% AR 10% 8 | 4, 1 en menor medida. | Fenotipo clínico. Biopsia. Genético | Mayores de 50 años. Ptosis de larga data con poco compromiso ocular, disfagia. |
| Miopatías distales | AD o AR (depende de subtipo) | 3 | RM muscular. Biopsia, Genético. | Diagnóstico diferencial con neuropatías. |
| Miopatias miofibrilares | Mayormente AD | 1,3,5,6 | RM muscular. Biopsia (depósitos miofibrilares). Genético | Amplio espectro fenotípico. Compromiso respiratorio y cardíaco. |
AD: Autosómica dominante; AR: Autosómica recesiva.
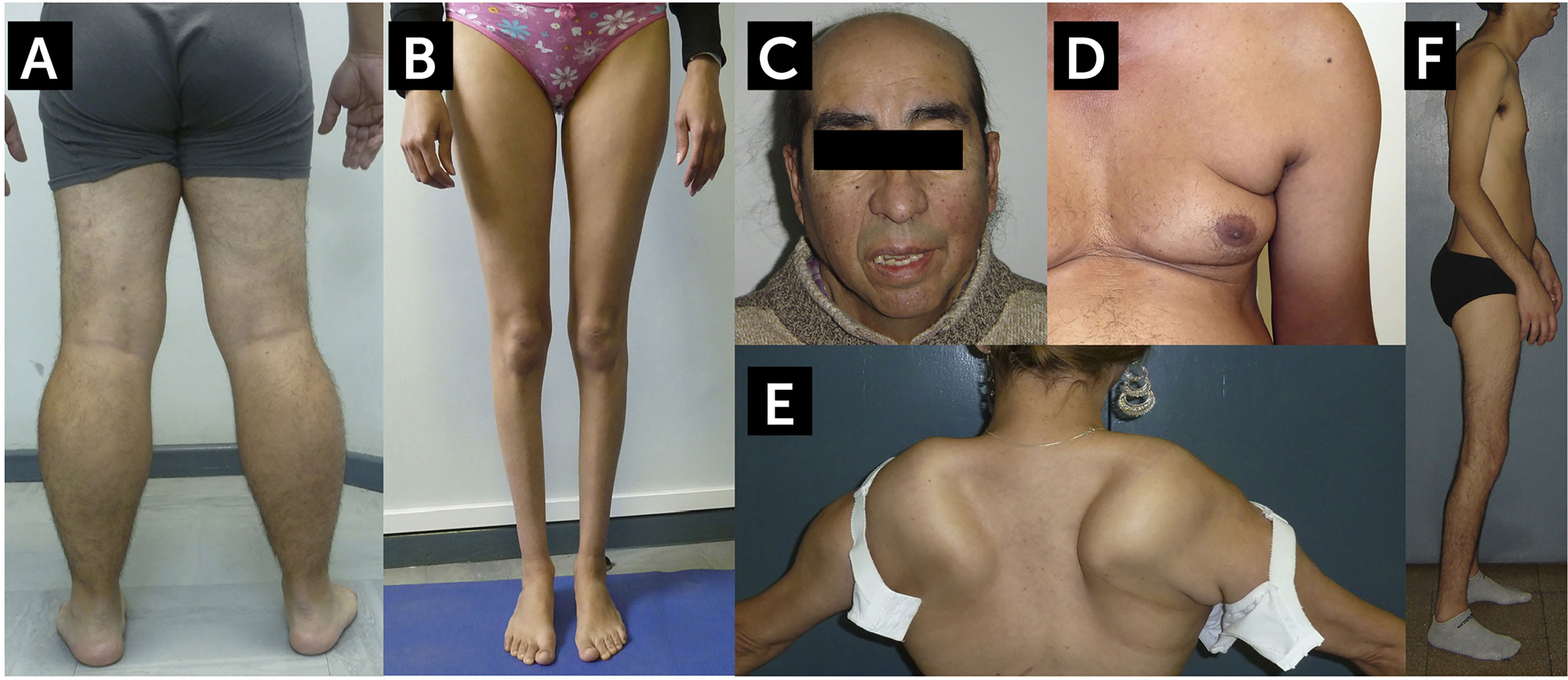
Distrofia muscular de Becker (DMB): Al igual que la DMD, la DMB es causada por una mutación ligada al X en el gen de la distrofina1,3,4. En la DMB, la mutación causal no altera el marco de lectura de la secuencia genética, produciendo una proteína de menor peso molecular parcialmente funcional, aunque excepcionalmente puede ser de mayor peso en caso de duplicaciones, llevando a un fenotipo más benigno, mientras que en la DMD la distrofina está ausente4. En la DMD se ha descrito clásicamente la pérdida de la deambulación antes de los 13 años, y después de los 16 años en la DMB, habiendo fenotipos intermedios si la pérdida de la deambulación ocurre entre los 13-16 años. Hoy se reconoce que la DMB tiene una edad de presentación variable, entre los 7 años en casos más severos, y después de los 30 años en los leves, algunos preservando deambulación sobre los 60 años4. Asimismo, el abordaje terapéutico actual de las distrofinopatías con corticoides y terapias nóveles, ha hecho que los pacientes con DMD lleguen a adultos28,29. Los pacientes presentan el patrón de debilidad tipo 1, con afección de cintura pélvica y en cuádriceps e isquiotibiales, pseudohipertrofia de las pantorrillas y acortamiento del tendón de Aquiles (Figura 1A)1. La mayoría se complica con cardiomiopatía y defectos de conducción, los que pueden ser la única manifestación de la enfermedad, y muchos pacientes desarrollan insuficiencia respiratoria. La CK se encuentra muy elevada, excepto en casos muy avanzados. La biopsia muscular muestra un patrón distrófico clásico, y se observa reducción parcial de la distrofina (uno o más de los antígenos que se deben explorar por inmunohistoquímica (IHQ) muestra una reacción parcial o incompleta), aunque en ocasiones la distrofina puede ser normal30. Las mujeres portadoras usualmente son asintomáticas aunque pueden presentar signos y síntomas (portadoras sintomáticas)4. Los hallazgos van desde hiperCKemia asintomática hasta un fenotipo similar al de un varón. El diagnóstico diferencial más importante de la DMB, incluye las sarcoglicanopatias (LGMD2C-F) y las mutaciones en el gen de la proteína relacionada con fukutina, FKRP (fukutin related protein) (LGMD2I)1,9.
. Vista posterior de las extremidades inferiores de un paciente con BMD. Se observa una marcada pseudohipertrofia de pantorrillas. B. Disferlinopatía, Miopatía de Miyoshi/LGMD2B. Se observa una severa atrofia de las pantorrillas. C. Facies Miotónica en un paciente con Distrofia Miotónica Tipo 1 (DM1, Enfermedad de Steinert). El paciente muestra calvicie, atrofia de músculos temporales, ptosis palpebral bilateral y paresia facial. Este paciente presentaba además un síndrome de cabeza caída por paresia extensora del cuello. D. Atrofia de la musculatura escapular en un caso de distrofia facio-escápulo-humeral (FSHD). Se observa el signo del pliegue axilar, abultamiento abdominal, y la atrofia del brazo. E. Despegamiento escapular en un caso de FSHD. F. Síndrome de Emery-Dreifuss en un caso de laminopatía. En el contexto de una amiotrofia generalizada, paciente presenta retracciones de codos, cuello y columna, la retracción Aquiliana está corregida por una intervención quirúrgica (tenotomía). Fotos autorizadas para publicación.")
Hallazgos característicos de algunas formas comunes de distrofia muscular del adulto
A. Distrofinopatía, distrofia de Becker (BMD). Vista posterior de las extremidades inferiores de un paciente con BMD. Se observa una marcada pseudohipertrofia de pantorrillas. B. Disferlinopatía, Miopatía de Miyoshi/LGMD2B. Se observa una severa atrofia de las pantorrillas. C. Facies Miotónica en un paciente con Distrofia Miotónica Tipo 1 (DM1, Enfermedad de Steinert). El paciente muestra calvicie, atrofia de músculos temporales, ptosis palpebral bilateral y paresia facial. Este paciente presentaba además un síndrome de cabeza caída por paresia extensora del cuello. D. Atrofia de la musculatura escapular en un caso de distrofia facio-escápulo-humeral (FSHD). Se observa el signo del pliegue axilar, abultamiento abdominal, y la atrofia del brazo. E. Despegamiento escapular en un caso de FSHD. F. Síndrome de Emery-Dreifuss en un caso de laminopatía. En el contexto de una amiotrofia generalizada, paciente presenta retracciones de codos, cuello y columna, la retracción Aquiliana está corregida por una intervención quirúrgica (tenotomía).
Fotos autorizadas para publicación.
Los pilares del manejo son la fisioterapia, la prevención y tratamiento de las complicaciones ortopédicas y de las complicaciones cardíacas4,23. El monitoreo cardíaco periódico es obligatorio en estos pacientes dado la alta tasa de muerte súbita. Los betabloqueadores e inhibidores de la angiotensina han mostrado beneficios4,24. La posibilidad de trasplante cardíaco es una alternativa que debe ser evaluada individualmente en cada caso24.
Si bien los glucocorticoides han demostrado claros beneficios en la DMD, en la DMB solo hay reportes de casos4,28. Algunos autores los usan en pacientes jóvenes con el fin de preservar la deambulación. La terapia génica está emergiendo como una estrategia válida y recientemente la FDA aprobó el eteplirsen, un oligonucleótido antisentido que trata la distrofia de Duchenne con mutaciones susceptibles al salto del exón 51 (13%)29. Existen otras estrategias en investigación, pero todavía no hay terapias aprobadas para la DMB.
Distrofias musculares de cinturas (LGMD): Las LGMD son grupo heterogéneo de miopatías primarias que se clasifican de acuerdo su forma de herencia en tipo 1 si es dominante, y Tipo 2 si es recesivo1,9. Se les asigna una letra en orden alfabético de acuerdo a la cronología del descubrimiento del gen causal. Esta clasificación ha sido recientemente revisada y actualizada31. En total abarcan alrededor de 30 enfermedades, cada una causada por mutaciones que afectan diferentes proteínas de la fibra muscular, pero que comparten ciertas características fenotípicas comunes1,9. Clínicamente siguen el patrón 1 de presentación, con algunas excepciones. La estrategia diagnóstica es intentar determinar la forma de herencia e identificar elementos distintivos. En el caso de las dominantes, si el patrón es escapuloperoneal (patrón 2) con retracciones, sospechar laminopatía A/C, si hay fenómeno rippling, caveolinopatía (LGMD1C). En las formas recesivas por ejemplo, si hay escápula alada, algunas contracturas y ausencia de compromiso cardíaco, y la biopsia no tiene hallazgos específicos, sospechar calpainopatía (LGMD2A); en cambio si la debilidad es distal (Figura 1B) en particular del compartimiento posterior de las piernas, con incapacidad de caminar en punta de pies, con CK muy elevadas, se debe sospechar disferlinopatía o anoctaminopatía (LGMD2B y L respectivamente)32. En estas formas, no se ha descrito compromiso facial, respiratorio, ni cardíaco. En casos que se presentan de forma similar a las distrofinopatías (DMD/DMB), particularmente en niños y niñas en la primera década de vida, con CK elevadas deben considerarse la LGMD2I (FKRP) y sarcoglicanopatías (LGMD2 C-F)32.
La biopsia muscular es de mucha ayuda en las LGMD, ya que con el uso de técnicas inmunohistoquímicas permite evidenciar en muchos casos la ausencia o disminución de la proteína mutada1,9,32,33. Se debe considerar que existen déficits secundarios de algunas proteínas como la distrofina, que pueden sugerir un diagnóstico erróneo si el panel de análisis no es completo, ya que producen una marcación irregular del antígeno33. Las distrofias con ausencia o déficit de alfa distroglicano son más comúnmente secundarias a mutaciones en proteínas que intervienen en su glicosilación, como son la fukutina o la proteína relacionada con fukutina (FKRP). Las llamadas distroglicanopatías incluyen la LGMD2I, 2K, 2M-O y la LGMD2P, esta última causada por mutaciones en el gen que codifica para el distroglicano alfa (DAG1), y también pueden manifestarse como distrofias musculares congénitas, de presentación pediátrica. La biopsia muscular de los pacientes con LGMD puede evidenciar la presencia de vacuolas o acumulación de proteínas como ocurre la titinopatía (LGMD2J), planteando el diagnóstico diferencial con miopatías distales o miofibrilares9,27. La RM muscular de cuerpo completo es una herramienta útil en el diagnóstico diferencial de las miopatías en general, incluyendo las LGMDs, y se han ido creando algoritmos para las distintas categorías de miopatía que ayudan a acotar el diagnóstico diferencial y en la selección de un músculo para biopsiar34,35. El diagnóstico definitivo es con el análisis genético molecular. El tratamiento es de soporte. Existe una herramienta online provista por le Fundación Jain Inc. dedicada al estudio de las disferlinopatías, que ayuda con el diagnóstico diferencial de las distintas distrofias en cinturas https://jain-foundation.org/alda/content/login-tool así como guías para su identificación y tratamiento32.
Distrofia miotónica: Existen dos formas de distrofia miotónica, tipo 1 y tipo 2 (DM1 y DM2 respectivamente)2,36. Nos enfocaremos en la primera que es la forma más común. Esta miopatía es autosómica dominante y se debe una expansión de los nucleótidos CTG en el gen DMKP2,13,36,37. La presentación va desde una forma congénita severa con muerte precoz, hasta fenotipos leves que se inician después de los 40 años y con una expectativa de vida normal, incluyendo todo el espectro de gravedad entre medio. La severidad depende de la longitud de la expansión de tripletes, y la expansión de los tripletes aumenta en cada generación de una familia afectada (fenómeno de anticipación)36. Esto hace que los hijos de un paciente afectado padezcan formas más severas de la enfermedad, la que a su vez será más severa en la generación siguiente. La forma clásica se presenta entre la segunda y cuarta décadas de la vida con dificultad para relajar la musculatura estriada, en particular en las manos, que tiende a mejorar con el ejercicio. La debilidad facial, mandibular y cervical, asociados a la calvicie frontal precoz, la atrofia del músculo temporal y la ptosis palpebral, le dan al paciente un aspecto muy característico que es típico de la enfermedad2,37. En las extremidades, la debilidad es de predominio distal (patrones 3 y 4). Los pacientes se quejan de disfagia, dificultad para usar sus manos, somnolencia diurna y a veces disnea, algunas veces presentándose con el patrón 5. Al examen se observa la calvicie característica, con ptosis palpebral simétrica, sin oftalmoplejía, ni diplopía y atrofia de los músculos temporales y maseteros, que determina un cierre bucal incompleto (facies miotónica, Figura 1C). La constatación de la miotonía de prensión y percusión confirman la sospecha clínica. La mayoría de los pacientes desarrolla cataratas precozmente y alteraciones cardiológicas que van desde trastornos de la conducción hasta cardiomiopat-a dilatada severa, siendo causa frecuente de morbimortalidad2,37. Existe además una asociación con alteraciones endocrinas como diabetes e hipogonadismo, molestias gastrointestinales por afectación del músculo liso del tubo digestivo y compromiso cognitivo disejecutivo e hipersomnolencia. La distrofia miotónica tipo 2 se presenta a edades más tardías, la miotonía clínica es mucho menos frecuente, la debilidad es proximal y el síntoma cardinal en muchos pacientes es el dolor muscular2,36,37.
El fenotipo es tan característico que a menudo se puede proceder al diagnóstico genético molecular sin necesidad de otros análisis. La alteración electromiográfica más prominente son las descargas miotónicas, aunque pueden estar ausentes en algunos casos. La demostración de la expansión de tripletes CTG en el gen DMPK por PCR es suficiente para establecer el diagnóstico36–38. En Chile, este análisis está disponible en la Clínica Alemana de Santiago y en la Red Salud UC.
La biopsia muscular no está indicada y sólo debe hacerse si existe una duda diagnóstica. El manejo se basa en el monitoreo de las complicaciones cardiológicas y respiratorias, que requiere la instalación de desfibriladores o marcapasos precozmente, así como de las otras manifestaciones multisistémicas de la enfermedad. Es necesario el tratamiento de la somnolencia, las apneas del sueño, así como el diagnóstico precoz y cirugía de las cataratas, endocrinopatías y disfagia. Para la miotonía severa la mexiletina es el fármaco de elección39.
Distrofia facioescapulohumeral (FSHD): La FSHD es una enfermedad autosómica dominante causada por deleción en tandem del gen D4Z4 que expresa la proteína DUX4 (FSHD tipo 1), y por mutaciones de SMCHD1 (FSHD tipo 2, 3% de los pacientes)5,40. La genética de esta enfermedad es compleja, heredándose de forma autosómica dominante en un 70-90% de los casos, pero puede originarse en mutaciones de novo hasta en un 30% de los casos40–42.
La FSHD es una miopatía característicamente asimétrica, clave importante en el diagnóstico. La debilidad afecta la cara, a veces de manera tan asimétrica que simula una parálisis del séptimo par craneano (patrón 2). El paciente tiene un cierre palpebral y del orbicularis oris incompletos, lo que le impide gesticular normalmente (Ej. silbar, hacer el gesto de un beso, sonreír). Se observa una escápula alada muy prominente y atrofia de la musculatura pectoral (pliegue axilar) y humeral (Figura 1D), con un respeto relativo del deltoides y de la musculatura antebraquial dando el aspecto de “brazo de Popeye”. La escápula puede protruir por sobre el trapecio dando una apariencia peculiar del hombro (Figura 1E). La musculatura abdominal baja se debilita, produciendo un desplazamiento del ombligo hacia cefálico al solicitarle al paciente que contraiga el abdomen (signo de Beevor). En las extremidades inferiores es típico observar debilidad de la dorsiflexión del pie, y pseudohipertrofia de pantorrillas, además de la atrofia de los cuádriceps. Hay variantes atípicas como el patrón escápulo-peroneal con respeto de la cara, la atrofia monomiélica, miopatía axial, distales y variantes infantiles, así como formas limítrofes40. En la mayoría de los casos la progresión es lenta permitiendo a los pacientes desempeñarse sin dificultad por largos períodos y, típicamente, no hay compromiso cardiorespiratorio. El diagnóstico es clínico ya que el cuadro es característico y se confirma con el análisis genético. El estudio electrofisiológico y la biopsia muscular no aportan información adicional en el algoritmo de estudio, por lo que, no están indicados a menos que haya una duda diagnóstica.
Distrofia de Emery Dreifuss (EDMD): Existe una variante ligada al X asociada a mutaciones de la emerina y variantes autosómicas dominantes asociadas a la lamina A/C, además de otros genes causantes menos frecuentes6,7. El patrón de compromiso es el escápulo-peroneal, con retracciones en codos que adoptan una postura en semiflexión y del talón de Aquiles (patrón 2, Figura 1F). Característicamente tienen cardiopatía, causa principal de morbimortalidad, lo que requiere vigilancia y tratamiento activo. La biopsia muscular tiene un patrón distrófico inespecífico. En los casos de mutaciones en el gen de emerina, se demuestra la ausencia de la proteína a nivel nuclear por inmunohistoquímica tanto en el músculo como en otros tejidos.
Distrofia oculofaríngea (OPMD): Es una miopatía poco frecuente, de presentación después de los 45 años, se caracteriza por la peculiar distribución de la debilidad (patrón 4)1,8,12. Los pacientes presentan ptosis progresiva, típicamente de larga data y que puede ser constatada en fotos antiguas. Es común que consulten cuando requieren operarse de la blefaroptosis. Los músculos extraoculares se encuentran habitualmente respetados en los estadios iniciales, lo que lo diferencia de las miopatías mitocondriales y la miastenia gravis. Junto con lo anterior, desarrollan disfagia neuromuscular y debilidad proximal de cinturas que va progresando con los años. La mayoría es autosómico dominante por expansión del triplete GCG en el gen PABPN19. En Latinoamérica se ha descrito una alta prevalencia y un efecto fundador en Uruguay12. En Chile hemos identificado varias familias con esta condición, aunque es probable que la enfermedad esté la subdiagnosticada17.
Otras miopatías relacionadas:Existe una larga lista de enfermedades musculares de presentación en el adulto43, algunas de las cuales no son clasificadas como distrofias, pero que deben ser correctamente diagnosticadas y diferenciadas de las distrofias antes descritas. El análisis detallado de cada una de ellas excede el propósito de la presente revisión, pero es importante saber de su existencia.
Las alteraciones del colágeno VI26 son formas relativamente frecuentes44 de distrofia congénita que se inician en la infancia en sus formas recesivas, más severas (miopatía de Ullrich), o pueden manifestarse de forma menos grave durante la primera o segunda décadas (síndrome de Bethlem), usualmente autosómica dominante. Se diferencian de otras distrofias, por la presencia de hiperlaxitud en los primeros años de vida, que evoluciona a retracciones y alteraciones cutáneas características, principalmente queloides25. Los pacientes mantienen su capacidad de deambular y usualmente no se acompañan de afección cardíaca. El diagnóstico se basa en la clínica, la RM muscular, que muestra un patrón de alteración característico y el estudio genético25. Es posible demostrar la alteración en la síntesis de colágeno en cultivos de fibroblastos a partir de la biopsia de piel o músculo del paciente, o se puede realizar el análisis genético a partir de fibroblastos cultivados, que permite una mejor caracterización del procesamiento molecular del colágeno alterado.
Las miopatías distales son un grupo poco frecuente de miopatías27 que se presentan con el patrón 3. Si bien algunas de ellas han sido clasificadas como distrofias de cinturas dominantes o recesivas (LGMD1E, LGMD2B), han sido ahora excluidas de esta categoría31. En casos de atrofia y debilidad de los gastrocnemios debe sospecharse una disferlinopatía o una anoctaminopatía (LGMD2B y 2L respectivamente)9,18, estos cuadros son frecuentemente confundidos con radiculopatías lumbosacras. Cuando la debilidad afecta el compartimiento anterolateral de la pierna, los pacientes se presentan con un síndrome de pie caído y una marcha en stepagge característica, que a menudo es erróneamente interpretada como una neuropatía18. En estos casos, el estudio electrofisiológico es importante para establecer la normalidad de la conducción nerviosa, con signos miopáticos en la electromiografía de aguja (irritabilidad de inserción, fibrilaciones, ondas agudas positivas, con trazados de esfuerzo de reclutamiento precoz y baja amplitud). Otras afecciones como la titinopatía y la desminopatía pueden igualmente presentarse con pie caído. Un grupo heterogéneo de estas miopatías distales están definidas por presentar acúmulos de proteínas citoesqueléticas y sarcoméricas en la biopsia muscular (“miopatías miofibrilares”), pueden transmitirse de forma autosómica dominante o recesiva y se presentan con los patrones clínicos 1, 3, 5 y 6. En éstas, el compromiso cardiorespiratorio es común. Una de las formas más comunes es la desminopatía45.
DIAGNÓSTICO DIFERENCIALEl diagnóstico diferencial de las miopatías en general es siempre dificultoso, por ser enfermedades poco frecuentes y por la gran sobreposición clínica que existe entre ellas. La anamnesis, la historia familiar en busca de un posible patrón de herencia y un examen físico acuciosos son las herramientas más determinantes para orientar correctamente el diagnóstico y el estudio complementario. Establecida la presunción de una miopatía, debe considerarse la posibilidad de una distrofia muscular frente a un cuadro clínico de debilidad muscular progresiva e indolora, que cursa con niveles elevados de creatina quinasa muscular (CK), a menudo con cambios tróficos (atrofia/pseudohipertrofia) en segmentos musculares específicos. En ese contexto, los diagnósticos diferenciales más relevantes en el adulto son las miopatías inflamatorias autoinmunes en sus distintas formas de presentación46, algunas formas de miopatía metabólica por acúmulo de glicógeno o trastornos del metabolismo lipídico, así como otras formas de miopatías no distróficas como por ejemplo las miopatías congénitas, tóxicas o infecciosas. Las canalopatías que cursan con debilidad y elevación de la CK, pueden confundirse tanto con una distrofia miotónica como con una distrofia muscular en su etapa inicial, pero típicamente son gatilladas por frío, potasio o ejercicio, lo que permite sospecharlas desde la anamnesis. En estos casos, la electrofisiología y el perfil evolutivo del cuadro son fundamentales para diferenciarlas. Los cuadros que cursan con debilidad pura, de curso insidioso y progresivo, pueden confundirse con una distrofia en ausencia de hallazgos específicos. En el adulto, hay que considerar entre ellos, los trastornos de las neuronas motoras como la esclerosis lateral amiotrófica, la enfermedad de Kennedy y la atrofia muscular espinal de presentación tardía (tipo III y IV). Los síndromes miasténicos autoinmunes (miastenia gravis, LEMS) y en particular algunas formas de miastenia congénita pueden remedar clínicamente una distrofia muscular. En pacientes en que la miopatía se ve en el contexto de un síndrome neurológico multisistémico, sordera, epilepsia, baja estatura, endocrinopatía y en particular si hay oftalmoplejía, hay que sospechar una miopatía mitocondrial47. Cuando una miopatía congénita se presenta después de la primera década de vida, entra también en el diagnóstico diferencial de una distrofia muscular del adulto. Una miopatía esporádica que puede confundirse con una distrofia muscular en el adulto es la miopatía por cuerpos de inclusión, que se presenta mayoritariamente en hombres después de los 50 años con disfagia, debilidad asimétrica de los flexores de los dedos, cuádriceps y tibial anterior, y que puede cursar con elevación de la CK (Tabla 3)46.
Algunas pautas para el diagnóstico diferencial de las DM en el adulto
| Polineuropatías | Compromiso distal, alteraciones sensitivas, pérdida de los reflejos, alteraciones en la velocidad de conducción. CK normal. |
| Enfermedad de Motoneurona (por ejemplo esclerosis lateral amiotrófica) | Distribución metamérica de la atrofia y la debilidad, hallazgos neurogénicos en la EMG, CK típicamente menor a 1000 UI/L. |
| Trastornos de la unión neuromuscular | Debilidad fluctuante, alteración electrofisiológica de la transmisión NM (decremento/potenciación del potencial motor), alteración de la EMG de fibra única (no es específica). |
| Miopatías adquiridas | Ausencia de historia familiar, exposición a medicamentos/tóxicos, alteraciones en parámetros metabólicos/endocrinológicos. |
| Presencia de otras enfermedades autoinmunes y /o sistémicas. | |
| Anticuerpos asociados a miositis o miositis específicos, biopsia compatible. | |
| Miositis por cuerpos de inclusión | Mayores de 50 años, compromiso selectivo de cuádriceps y flexores de dedos, CK relativamente baja, biopsia compatible, expresión normal de proteínas en la biopsia. |
| Miopatías metabólicas | Intolerancia al ejercicio, mialgias relacionadas al ejercicio, mioglobinuria/rabdomiolisis a repetición, fenómeno del segundo aliento. Biopsia con depósitos de glicógeno o lípidos. |
| Miopatías mitocondriales | Herencia materna, compromiso multisistémico asociado: epilepsia, baja estatura, sordera sensorio-neural, oftalmoplejia externa, cardiomiopatía, retinitis pigmentosa, lactato incrementado, diabetes. Biopsia compatible. |
EMG: Electromiografía; CK: creatinina quinasa.
Como se indicó más arriba, las distrofias musculares usualmente cursan con elevación de la CK. Los niveles de CK muy elevados se observan en las distrofinopatías, disferlinopatías, caveolinopatías y sarcoglicanopatías, aunque puede estar en rango normal o levemente elevadas en la FSHD, DM1, OPMD y en el síndrome de Emery-Dreifuss. La elevación concurrente de las transaminasas séricas GOT, GPT y de LDH como consecuencia de la destrucción muscular (y no por afección hepática) orienta a un proceso muscular distrófico o inflamatorio. Una distrofia muscular puede debutar con un episodio de rabdomiolisis en el contexto de actividad física o espontáneamente. Los restantes análisis de laboratorio son útiles para descartar otros diagnósticos diferenciales, pero no se alteran específicamente en la distrofia muscular. En cuadros de inicio agudo o subagudo, los anticuerpos músculo-específicos o asociados a miositis (panel miositis) y los anticuerpos anti-HMGCR, son útiles en el diagnóstico diferencial de las distrofias con las miopatías inflamatorias, los que pueden estar también elevados en niños con un fenotipo parecido a una distrofia inicial48. Estos son específicos de los procesos autoinmunes y no suelen elevarse en las distrofias musculares, así como tampoco otros parámetros inflamatorios.
El estudio electrofisiológico no permite un diagnóstico positivo de una distrofia muscular. En el mejor de los casos confirma la presencia de un trastorno miopático y/o su distribución, aunque puede resultar completamente normal. Su mayor valor reside en excluir ciertos diagnósticos diferenciales como por ejemplo una neuropatía, una enfermedad de las neuronas motoras o un trastorno de la unión neuromuscular. Debe hacerse la excepción de la distrofia miotónica y otros trastornos miotónicos, que sí presentan alteraciones características y a menudo diagnósticas. En general, las conducciones nerviosas se hallan normales o pueden mostrar una caída en la amplitud de los potenciales motores por atrofia del efector muscular, en ausencia de alteración de la conducción sensitiva. La electromiografía muestra hallazgos miogénicos que dependen del tiempo de evolución del proceso distrófico. En etapas precoces hay irritabilidad de membrana, ondas agudas positivas, fibrilaciones, asociados en diferente proporción potenciales de esfuerzo de baja amplitud y reclutamiento precoz. Conforme avanza la destrucción muscular se sobre agregan cambios crónicos, que se entremezclan en distinta proporción con las alteraciones tempranas (ej. potenciales de gran amplitud, polifásicos, “pseudo-neurogénicos”). Dado que las distrofias afectan distintos músculos de forma asincrónica, esta secuencia de eventos puede darse en distintos tiempos en diferentes músculos, y en ese sentido la selección de músculos a explorar puede ser un factor importante en el resultado final del estudio.
La resonancia magnética (RM) muscular permite evaluar el cuerpo completo, caracterizando el trofismo muscular, el grado de reemplazo graso, la presencia de edema muscular, así como la eventual afección de otras vísceras (Ej. corazón, estómago)18,34,35. Se han creado diferentes algoritmos para los distintos tipos de miopatías incluyendo las distrofias, que ayudan en el diagnóstico diferencial y ocasionalmente permiten orientar el estudio genético molecular32. Es útil además para escoger un músculo adecuado para la biopsia.
La biopsia muscular es fundamental en el diagnóstico de las distrofias. Confirma la presencia de la distrofia y permite además, a través de la inmunohistoquímica (IHQ), detectar la ausencia o reducción de proteínas causales de la misma, así como determinar la presencia de marcadores histológicos de otras miopatías que entran en el diagnóstico diferencial1,9,33. La presencia en la biopsia de un patrón distrófico con microscop-a de luz, es inespecífico y exige completar el análisis histológico con técnicas de IHQ32,33, aun así, algunas distrofias no presentan marcadores específicos y el diagnóstico final debe hacerse a través del análisis molecular de proteínas por western blot en músculo o sangre, o un estudio genético molecular32,33,49,50. La microscop-a electrónica no es útil en el diagnóstico positivo de las distrofias musculares, a excepción de la OPMD, pero sí en el caso de miopatías congénitas estructurales o en las miopatías miofibrilares, que pueden entrar en el diagnóstico diferencial de una distrofia.
El análisis genético es indispensable en diagnóstico de una distrofia muscular y debe confirmarse siempre con el análisis genético el gen causante de la misma. Cuando el fenotipo clínico es muy sugerente de un diagnóstico, el análisis genético molecular es la primera opción para confirmar la causa (Ej. BMD, DM1, FSHD, etc.). El estudio genético debe ser orientado a través un análisis fenotípico minucioso en el paciente y sus familiares directos, y de un correlato acucioso con todos los hallazgos clínicos y pruebas paraclínicas. La evaluación de familiares afectados no solo permite establecer más correctamente el patrón de herencia, sino que contribuye a la identificación de portadores asintomáticos, así como para realizar un asesoramiento genético adecuado para el paciente y su familia.
Si bien el uso de los paneles genéticos es muy útil y llega a resolver hasta un 50% de los diagnósticos en series de pacientes seleccionados, debe considerarse que ningún panel abarca la totalidad de las alteraciones genéticas posibles. En este sentido, su utilización no exime la necesidad del análisis del fenotipo clínico, y requiere de una evaluación cuidadosa post-análisis genético, para correlacionar los hallazgos con el fenotipo clínico, ya que en muchos casos se obtienen variantes de significado incierto de difícil interpretación49,50.
ABORDAJE TERAPÉUTICOEl pilar del tratamiento es el soporte1,3,32. Las unidades multidisciplinarias que integran a neurólogos, cardiólogos, fisiatras, kinesiólogos, traumatólogos, neumólogos, etc., obtienen los mejores resultados y permiten adelantar las complicaciones que van surgiendo. El primer paso de un abordaje terapéutico adecuado es tener un diagnóstico de certeza. Con frecuencia hay pacientes que han sido tratados por períodos prolongados con un diagnóstico erróneo, típicamente de miopatía inflamatoria y expuestos a tratamientos potencialmente nocivos, que no solo no favorecen el curso de la distrofia, sino que le agregan morbimortalidad. Los resultados con terapias específicas basadas en la fisiopatología molecular, como el uso de oligómero morfolino etplirsen que favorece el salto exónico del exón 51 del gen de distrofina29, o el reemplazo enzimático de la alfa glucosidasa A en la enfermedad de Pompe51 entre otras, que no eran imaginables pocos años atrás, permiten vislumbrar un futuro promisorio para el tratamiento específico de las miopatías en general.
CONCLUSIONESEn suma, las distrofias musculares son un grupo de enfermedades de presentación variable donde la debilidad de la musculatura estriada es el síntoma cardinal. La semiología sigue siendo fundamental en el proceso diagnóstico y nos permite establecer la edad de presentación, un patrón de herencia, localización de los grupos musculares afectados, la existencia de fenómenos como la miotonía y compromiso extramuscular. Estos datos nos permiten obtener un diagnóstico diferencial y establecer una probabilidad “pretest” (Ej. previa a la realización de un análisis molecular), para poder interpretar adecuadamente los resultados de las pruebas diagnósticas (“análisis post-test”). Las herramientas paraclínicas principales son la medición de CK, la biopsia muscular, la RM muscular y la genética. El tratamiento de soporte por un equipo multidisciplinario es de suma importancia para el manejo de estas enfermedades progresivas. Cada vez se hace más importante llegar al diagnóstico etiológico ya que están apareciendo tratamientos específicos modificadores de la enfermedad y además le permite a los pacientes de participar en ensayos clínicos. Por último hay que destacar que al ser enfermedades poco frecuentes se requerirá de un esfuerzo mancomunado a nivel mundial para poner a prueba las terapias del futuro.
Declaración de interésLos autores de este artículo declaran no presentar ningún conflicto de interés.
FinanciamientoProyecto FONDECYT 1151383 aJB.