



Las miopatías inflamatorias son un grupo heterogéneo de enfermedades adquiridas del músculo estriado esquelético que comparten la injuria muscular inmunomediada como característica común. En esta revisión se repasarán las principales formas de miopatía inflamatoria, con énfasis en las miopatías inflamatorias idiopáticas.
The inflammatory myopathies are a heterogeneous group of acquired muscle disorders, which share the common feature of immune mediated muscle injury. In this review, the main forms of inflammatory myopathy will be discussed, with emphasis on the idiopathic inflammatory myopathies.
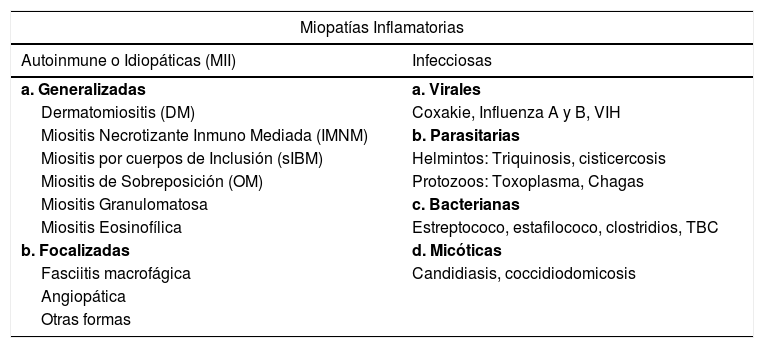
Las miopatías inflamatorias (MI) o miositis1, son un grupo heterogéneo de enfermedades musculares de rara ocurrencia, caracterizadas por la inflamación de los distintos componentes del tejido muscular, ya sea de forma aislada o más comúnmente en el contexto de una afección sistémica. Más allá de ciertas similitudes clínicas e histopatológicas, el espectro de las MI es considerablemente variable. La biopsia muscular en MI típicamente muestra infiltrados inflamatorios de distinta magnitud, aunque estos pueden estar ausentes, dependiendo del momento evolutivo de la enfermedad o el tipo de miositis, sin olvidar que en las miopatías hereditarias también pueden haber cuadros histológicos indistinguibles2. El diagnóstico de una MI requiere de la exclusión de otras formas de miopatía que remedan una MI incluyendo las distrofias de cinturas, la distrofia facio-escápulo-humeral, así como miopatías metabólicas, mitocondriales, endocrinas e inducidas por drogas3,4. La identificación correcta del subtipo de MI es fundamental para establecer una correcta estrategia de estudio, determinar el pronóstico e implementar el tratamiento adecuado4. Las miopatías inflamatorias pueden dividirse en aquellas que tienen causa conocida y las primarias, de causa indeterminada o idiopáticas (MII)1 (Tabla 1).
Clasificación de las miopatías inflamatorias1
| Miopatías Inflamatorias | |
|---|---|
| Autoinmune o Idiopáticas (MII) | Infecciosas |
| a. Generalizadas | a. Virales |
| Dermatomiositis (DM) | Coxakie, Influenza A y B, VIH |
| Miositis Necrotizante Inmuno Mediada (IMNM) | b. Parasitarias |
| Miositis por cuerpos de Inclusión (sIBM) | Helmintos: Triquinosis, cisticercosis |
| Miositis de Sobreposición (OM) | Protozoos: Toxoplasma, Chagas |
| Miositis Granulomatosa | c. Bacterianas |
| Miositis Eosinofílica | Estreptococo, estafilococo, clostridios, TBC |
| b. Focalizadas | d. Micóticas |
| Fasciitis macrofágica | Candidiasis, coccidiodomicosis |
| Angiopática | |
| Otras formas | |
Ocasionalmente vemos que algunos pacientes presentan, en el contexto de una infección viral inespecífica por ejemplo, mialgias y elevación transitoria de enzimas musculares en la sangre, indicativos un proceso muscular inflamatorio. Estas formas secundarias de MI sólo ameritan un manejo sintomático. Sin embargo, otros pacientes pueden desarrollar un cuadro más grave y persistente causado por una enfermedad infecciosa y que requiere el tratamiento específico. Un ejemplo de MI de etiología conocida es la triquinosis. Una parasitosis sistémica provocada por el nematodo Trichinella spiralis, que coloniza al organismo al ingerir, más comúnmente, carne de cerdo contaminada con larvas del parásito5. Los pacientes desarrollan una polimiositis parasitaria con los signos y síntomas propios de la inflamación muscular, asociados de forma variable a un compromiso sistémico de distinta gravedad, los que revierten al ser tratados efectivamente con antiparasitarios.
Sin embargo, de forma más común actualmente, la MI se presenta en ausencia de una causa identificable, haciendo plantear la existencia de una enfermedad primaria e idiopática del músculo3,6. Estas miopatías inflamatorias idiopáticas (MII) son generalmente inmunomediadas, y suelen ocurrir en el contexto de una afección multisistémica, asociado a otras enfermedades autoinmunes, a la presencia de cáncer (paraneoplásico) o gatilladas por el uso de medicamentos miotóxicos, como es el caso de las estatinas2,3,6. En las MII la identificación de anticuerpos asociados o específicos, en conjunto con la biopsia muscular y otros métodos complementarios de estudio, son muy relevantes para establecer el diagnóstico, el pronóstico y el tratamiento, además de permitir una clasificación de las MI basadas en la ocurrencia de marcadores biológicos2,7.
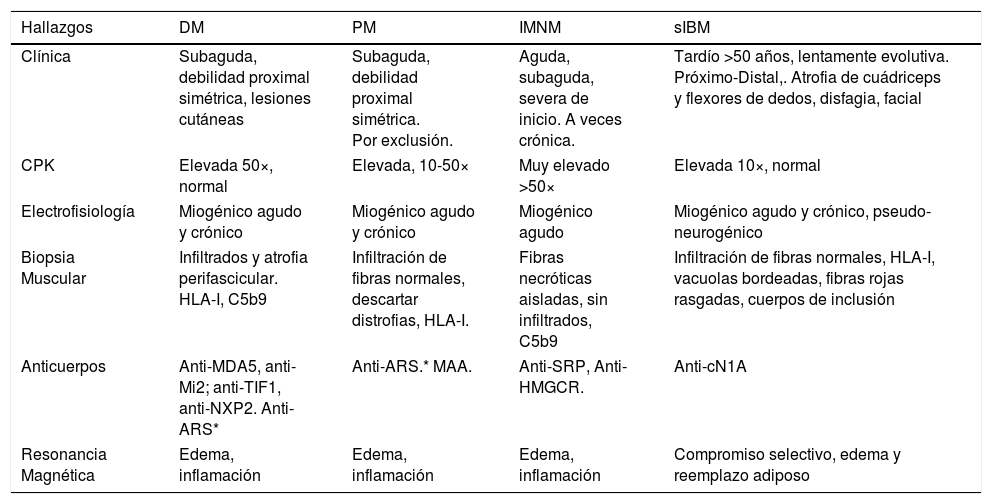
En esta revisión nos enfocaremos en este último grupo de MI, las miopatía inflamatorias idiopáticas, que incluye la dermatomiositis (DM), la polimiositis (PM), la miositis necrotizante inmuno mediada (IMNM), las miositis de sobreposición (OM) y la miositis por cuerpos de inclusión (IBM) (Tabla 2)2,4–6.
Principales diferencias entre las distintas formas de MII
| Hallazgos | DM | PM | IMNM | sIBM |
|---|---|---|---|---|
| Clínica | Subaguda, debilidad proximal simétrica, lesiones cutáneas | Subaguda, debilidad proximal simétrica. Por exclusión. | Aguda, subaguda, severa de inicio. A veces crónica. | Tardío >50 años, lentamente evolutiva. Próximo-Distal,. Atrofia de cuádriceps y flexores de dedos, disfagia, facial |
| CPK | Elevada 50×, normal | Elevada, 10-50× | Muy elevado >50× | Elevada 10×, normal |
| Electrofisiología | Miogénico agudo y crónico | Miogénico agudo y crónico | Miogénico agudo | Miogénico agudo y crónico, pseudo-neurogénico |
| Biopsia Muscular | Infiltrados y atrofia perifascicular. HLA-I, C5b9 | Infiltración de fibras normales, descartar distrofias, HLA-I. | Fibras necróticas aisladas, sin infiltrados, C5b9 | Infiltración de fibras normales, HLA-I, vacuolas bordeadas, fibras rojas rasgadas, cuerpos de inclusión |
| Anticuerpos | Anti-MDA5, anti-Mi2; anti-TIF1, anti-NXP2. Anti-ARS* | Anti-ARS.* MAA. | Anti-SRP, Anti-HMGCR. | Anti-cN1A |
| Resonancia Magnética | Edema, inflamación | Edema, inflamación | Edema, inflamación | Compromiso selectivo, edema y reemplazo adiposo |
(*) Pueden estar elevados DM y PM. MAA: anticuerpos asociados a miositis. Ver también tabla 3.
CK: creatinina quinasa.
Ref. 2,4-,6.
El objetivo principal de este artículo es dar una visión general actualizada de las formas clínicas, los criterios de clasificación, la asociación de las distintas formas con presencia de niveles aumentados de anticuerpos y los lineamientos generales del diagnóstico, el manejo y tratamiento.
EPIDEMIOLOGÍALas MI son en conjunto enfermedades raras con una incidencia aproximada de 1 caso cada 100000 individuos, pero constituyen el grupo más grande de miopatías potencialmente tratables en niños y adultos2. Si bien entre las enfermedades neuromusculares las MII son más frecuentes, siguen siendo enfermedades raras por su baja incidencia 11/1000000/año y prevalencia 14/1000008. Con excepción de la dermatomiositis juvenil9, las MII son enfermedades que afectan mayormente a adultos 6, aunque la presentación en niños está siendo comunicada con mayor frecuencia en los últimos dos años10,11. La edad de inicio de las MII está en entre los 45–60 años en adultos, y alrededor de los 15 años para las formas juveniles12. Afectan más a mujeres que hombres en proporción 2:1, con excepción de la miopatía por cuerpos de inclusión (IBM), donde la relación hombre/mujer puede llegar a 3:112. La prevalencia ajustada por edad de la IBM, oscila entre 50.1 y 70 casos por millón, y hace que esta sea la MII más frecuente en mayores de 50 años13,14. Las miopatías necrotizanes representan el 19% de las MII, mientras que la DM y las miositis inespecíficas dan cuenta del 36% y 39% de todas las MII respectivamente6,15. La polimiositis (PM) por otra parte, ha sido cuestionada como una entidad nosológica y en algunos estudios ha sido encontrada con mucho menos frecuencia de lo estimado dando cuenta de sólo el 2% de los casos15.
CLASIFICACIÓN DE LAS MIOPATÍAS INFLAMATORIAS IDIOPÁTICASSi bien la existencia de enfermedades inflamatorias del músculo ha sido reconocida desde hace mucho tiempo atrás 16, el conocimiento de sus manifestaciones clínicas e histopatológicas, así como del manejo y tratamiento de las mismas, han cambiado dramáticamente desde la publicación de los criterios diagnósticos de Bohan y Peter en 197517,18, que han sido los más usados desde entonces. Si bien prácticos en general, los criterios de Bohan y Peter presentan algunos inconvenientes importantes. Por un lado son demasiado inclusivos, es decir, caen dentro del criterio de “miositis” enfermedades que no lo son como por ejemplo distrofias musculares o enfermedades metabólicas; y en segundo lugar excluyen enfermedades que si son de naturaleza inflamatoria como la miopatía por cuerpos de inclusión o la miopatía necrotizante autoinmune, además de no tomar en consideración la serología y los mecanismos fisiopatogénicos. El descubrimiento de nuevos métodos de diagnóstico y de una serie de autoanticuerpos que son específicos (myositis specific antibodies, MSA) o se asocian (myositis associated antibodies, MAA) con distintas subformas de MII7 (Tabla 3), ha dado lugar a criterios diagnósticos consensuados para una nueva clasificación basada en los hallazgos clínicos, serológicos e histopatológicos16,19,20. Estos criterios son el resultado de una colaboración internacional multidisciplinaria; se basa en los datos objetivos e incluye definiciones de las variables a considerar21. De acuerdo con este enfoque, el espectro clínico de las MII ha evolucionado desde enfermedades principalmente musculares y cutáneas, a condiciones inflamatorias sistémicas con afectación multiorgánica, a menudo grave y fatal16.
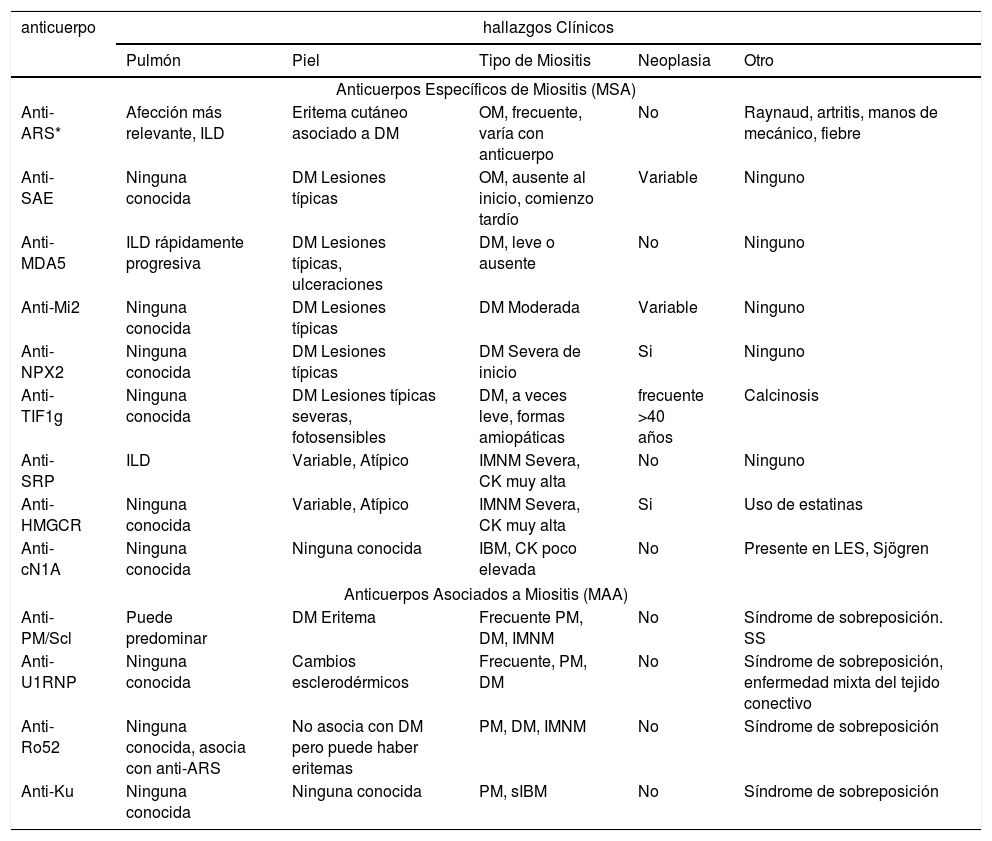
Anticuerpos específicos y asociados a Miositis7,33
| anticuerpo | hallazgos Clínicos | ||||
|---|---|---|---|---|---|
| Pulmón | Piel | Tipo de Miositis | Neoplasia | Otro | |
| Anticuerpos Específicos de Miositis (MSA) | |||||
| Anti-ARS* | Afección más relevante, ILD | Eritema cutáneo asociado a DM | OM, frecuente, varía con anticuerpo | No | Raynaud, artritis, manos de mecánico, fiebre |
| Anti-SAE | Ninguna conocida | DM Lesiones típicas | OM, ausente al inicio, comienzo tardío | Variable | Ninguno |
| Anti-MDA5 | ILD rápidamente progresiva | DM Lesiones típicas, ulceraciones | DM, leve o ausente | No | Ninguno |
| Anti-Mi2 | Ninguna conocida | DM Lesiones típicas | DM Moderada | Variable | Ninguno |
| Anti-NPX2 | Ninguna conocida | DM Lesiones típicas | DM Severa de inicio | Si | Ninguno |
| Anti-TIF1g | Ninguna conocida | DM Lesiones típicas severas, fotosensibles | DM, a veces leve, formas amiopáticas | frecuente >40 años | Calcinosis |
| Anti-SRP | ILD | Variable, Atípico | IMNM Severa, CK muy alta | No | Ninguno |
| Anti-HMGCR | Ninguna conocida | Variable, Atípico | IMNM Severa, CK muy alta | Si | Uso de estatinas |
| Anti-cN1A | Ninguna conocida | Ninguna conocida | IBM, CK poco elevada | No | Presente en LES, Sjögren |
| Anticuerpos Asociados a Miositis (MAA) | |||||
| Anti-PM/Scl | Puede predominar | DM Eritema | Frecuente PM, DM, IMNM | No | Síndrome de sobreposición. SS |
| Anti-U1RNP | Ninguna conocida | Cambios esclerodérmicos | Frecuente, PM, DM | No | Síndrome de sobreposición, enfermedad mixta del tejido conectivo |
| Anti-Ro52 | Ninguna conocida, asocia con anti-ARS | No asocia con DM pero puede haber eritemas | PM, DM, IMNM | No | Síndrome de sobreposición |
| Anti-Ku | Ninguna conocida | Ninguna conocida | PM, sIBM | No | Síndrome de sobreposición |
(*) ARS (aminoacil-tARN sintetasa) se refiere a los anticuerpos conocidos como “antisintetasa”, e incluye: anti-Jo1 (anti-histidil-tARN sintetasa), anti-PL7 (anti-treonil-tARN sintetasa), anti-PL12 (anti-alanil-tARN sintetasa), anti-EJ (anti-glicil-tARN sintetasa), anti-OJ (anti-isoleucil-tARN sintetasa), anti-Ha (anti-tirosil-tARN sintetasa), anti-KS (anti-asparagil-tARN sintetasa) y anti-Zo (anti-fenilalanil-tARN sintetasa). ILD, enfermedad pulmonar intersticial, DM, dermatomiositis; IMNM, miopatía necrotizante inmuno-mediada; PM, polimiositis; OM, miositis de sobreposición; sIBM, miositis por cuerpos de inclusión. LES, lupus eritematoso sistémico; SS, esclerosis sistémica.
El detalle de la discusión que ha llevado a la clasificación actual de las MII está mas allá del alcance de esta revisión y ha sido extensamente revisado en publicaciones recientes16. Baste decir que actualmente las MIIs comprenden al menos cinco síndromes mayores: la dermatomiositis (DM), la polimiositis (PM), la miopatía necrotizante autoinmune (IMNM), la miopatía de sobreposición (OM), y la miositis por cuerpos de inclusión esporádica (IBM). Siendo esta última una entidad cuya naturaleza autoinmune está en discusión2,3.
Los nuevos criterios diagnósticos desarrollados por la EULAR,19,20 no solo permiten identificar e incluir en la clasificación pacientes sin manifestaciones musculares (Ej. dermatomiositis amiopática), sino que también dan relevancia a hallazgos clínicos característicos en ausencia de hallazgos histopatológicos, considerados excluyentes en los criterios previamente establecidos, como es el caso de la miositis por cuerpos de inclusión16.
PRESENTACIÓN CLÍNICA GENERALLas miopatías inflamatorias o miositis (MI) deben sospecharse en pacientes con un cuadro de debilidad presentación aguda (miopatías necrotizantes, DM), subaguda (DM, OM) o crónica (IBM, PM)3,4,6. La debilidad, de curso insidioso, afecta predominantemente la musculatura proximal de las cinturas pélvica y escapular de forma simétrica, pero también el cuello (síndrome de cabeza caída) y el tronco, en ausencia de oftalmoplejía y/o ptosis, y de síntomas sensitivos2,4. Los pacientes se quejan de dificultad para tareas cotidianas como elevar los brazos (maquillarse, peinarse), levantarse de una silla, caminar o subir escaleras, levantar cosas pesadas, y a veces de tareas que involucran la musculatura distal (manipular llaves, caminar en puntas y/o talones). La afección de la musculatura respiratoria y del diafragma puede llevar a insuficiencia respiratoria21. El compromiso bulbar se manifiesta por disfagia, disartria y a veces voz nasal24. La debilidad puede acompañarse de un compromiso del estado general, fatigabilidad, mialgias, artralgias, fenómeno de Raynaud, asociado o no a una respuesta inflamatoria sistémica con afectación multiorgánica, baja de peso y fiebre2,22,23. Algunas de esas manifestaciones son el compromiso respiratorio (enfermedad pulmonar intersticial, ILD)21,25, manifestaciones cutáneas26,27, la afección cardíaca (arritmias, cardiomiopatías) o la asociación con cáncer (paraneoplásica)22.
La miositis aislada o “pura”, sin manifestaciones extramusculares también puede ocurrir y obliga a establecer la eventual presencia de una condición causal de la misma. La MI habitualmente cursa con una elevación sérica de la creatina quinasa muscular (CK), de rango variable, pudiendo llegar a niveles que ocasionalmente provocan una falla renal por mioglobunuria, aunque puede no estar elevada. Suele haber una elevación concurrente de las transaminasas séricas GOT (AST), GPT (ALT) y de LDH como un indicador de la destrucción muscular (y no por afección hepática, la GGT que es producida específicamente por este órgano tiende a estar normal)3.
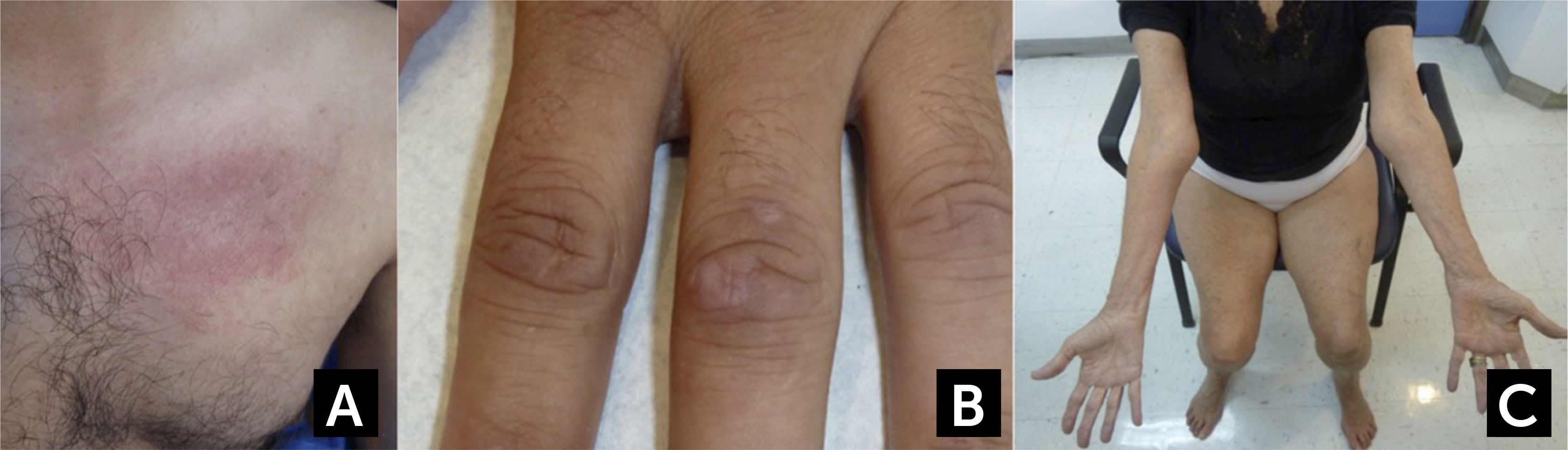
FORMAS CLÍNICAS ESPECÍFICASDermatomiositisEl rasgo distintivo de la DM es el compromiso de la piel, que se manifiesta como un exantema cutáneo el que puede preceder a la debilidad, o bien presentarse sin ella (dermatomiositis amiopática)28, aunque este puede presentarse de forma muy similar en el síndrome antisintetasa (ASS)7. Los pacientes muestran erupciones de color rojo-púrpura o violáceas en las áreas fotosensibles4,27,28. Típicamente se observan en los párpados y alrededor de las órbitas (eritema heliotropo), en el resto de la cara, en forma de V en la base del cuello, en el pecho y en la espalda superior y hombros (signo del chal) (Figura 1A). Las pápulas de Gottron se producen de forma simétrica sobre la región dorsal de las articulaciones metacarpofalángicas e interfalángicas, pero también pueden haber pápulas en las superficies extensoras de otras articulaciones como codos, rodillas o tobillos (Signo de Gottron) (Figura 1B). En la base de las uñas pueden presentarse capilares dilatados, cutículas “irregulares”, que suelen acompañarse de telangienctasias e infartos hemorrágicos en las zonas hipertróficas de la cutícula. Las calcificaciones subcutáneas (calcinosis cutánea) pueden ocurrir hasta en la mitad de los casos de DM juvenil severa23,26,27. En la DM la debilidad muscular es de inicio subagudo a nivel de cintura escapular y pélvica. Los pacientes suelen quejarse de mialgias. En algunos casos de presentación más aguda, la rabdomiolisis puede ser severa y provocar una falla renal por mioglobinuria. En un tercio de los casos suele haber disfagia, así como debilidad palpebral28. La dificultad para tragar y los signos de aspiración demuestran el compromiso de la musculatura estriada de la faringe y el tercio superior del esófago, cuando están presentes denotan un curso agresivo de la DM, que puede estar asociado con un mal pronóstico24,28. La debilidad puede comprometer a la musculatura respiratoria en los casos más severos. Las articulaciones, el tracto gastrointestinal, el pulmón (enfermedad intersticial) y el corazón (alteraciones de la conducción y miocardiopatía) pueden estar también presentes28.
 Rash eritematoso en la base del cuello y región pectoral en un caso de dermatomiositis. (B) Pápulas de Gottron en dermatomiositis. (C) Atrofia de los flexores largos de los dedos a nivel del antebrazo y de cuádriceps en un caso de miositis por cuerpos de inclusión esporádica. Fotos autorizadas por padres y/o pacientes.")
Características clínicas en dermatomiositis y Miositis por Cuerpos de Inclusión
(A) Rash eritematoso en la base del cuello y región pectoral en un caso de dermatomiositis. (B) Pápulas de Gottron en dermatomiositis. (C) Atrofia de los flexores largos de los dedos a nivel del antebrazo y de cuádriceps en un caso de miositis por cuerpos de inclusión esporádica.
Fotos autorizadas por padres y/o pacientes.
Los pacientes con dermatomiositis presentan elevación de anticuerpos específicos incluyendo anti-Mi2 que asocia con las típicas lesiones cutáneas; anti-MDA5, que asocia con lesiones cutáneas y enfermedad pulmonar intersticial; anti factor de trascripción intermediario 1γ (anti-TIF1γ) en pacientes que suelen presentar lesiones cutáneas severas y cáncer, y anti-proteína de la matriz nuclear 2 (anti-NXP2) que en pacientes adultos se asocia a cáncer. Es importante destacar que lesiones cutáneas características de DM, pueden también encontrarse en pacientes con síndrome antisientetasa (ASS) (ver más adelante), en los que la histopatología, así como la serología contribuyen a identificarlos7. La expresión de estos anticuerpos está igualmente influenciada por factores geográficos, raciales y genéticos3,7,22.
PolimiositisLa polimiositis PM23 es mayormente un diagnóstico de exclusión para todas las MII que no son DM o miopatía por cuerpos de inclusión esporádica (sIBM) y actualmente ha sido cuestionada como una entidad nosológica específica2,15,29. La enfermedad más comúnmente diagnosticada como PM es la sIBM, la que es sospechada retrospectivamente cuando el paciente no responde al tratamiento2,29. El uso todavía vigente de los criterios de Bohan y Peter17,18 para diagnosticar PM, ha inducido al diagnóstico erróneo de PM en casos de DM con escasas manifestaciones cutáneas, miopatías necrotizantes inmuno-mediadas (IMNM), síndromes de sobreposición (OM), distrofias musculares con inflamación u otras miopatías, por lo que su existencia como una entidad nosológica aislada ha sido relativizada la práctica actual4,30.
Miopatía Necrotizante Inmuno-Mediada (IMNM)La miopatía necrotizante inmunomediada (IMNM)3,4 es una entidad clínico-patológica que da cuenta de hasta el 19% de las MII15. Si bien puede ocurrir a cualquier edad, es más frecuente en adultos31 y cursa con una elevación muy marcada de la CK y de los títulos de MSA o MAA (Tabla 2). Puede presentarse de forma espontánea, aislada, o en el contexto de una infección viral, asociado a cáncer, en pacientes con enfermedades autoinmunes del tejido conectivo o expuestos a estatinas, aun después de suspendida su ingesta3,32. La presentación clínica de la IMNM depende de la causa que la determina. En su forma más típica el inicio es agudo y severo, llegando a su máxima expresión en días o semanas; menos frecuentemente es subagudo, progresando a una debilidad severa en el lapso de meses3. Aproximadamente un 60% de los pacientes con IMNM muestran títulos elevados de anticuerpos anti-SRP o anti-HMGCR3,31. Es interesante destacar que, al menos en teoría, las estatinas son la única droga capaz de generar una reacción autoinmune contra su propio blanco farmacológico. Los anti-HMGCR también se dan en pacientes no expuestos a estatinas y tienen presentación más temprana, menor respuesta a fármacos y curso más agresivo3. Descartada la posibilidad de una causa tóxica o relacionada a drogas, debe realizarse la medición de anticuerpos anti-SRP (“signal recognition particle) o anti 3-hidroxi-3-metilglutaril–coenzima A reductasa (anti-HMGCR) ya que si están elevados se confirma el diagnóstico de IMNM y valida el inicio de la terapia inmunosupresora. La titulación de otros MSA y MAA (panel de autoanticuerpos para miositis) permitirá establecer la presencia de otras formas de IMNM. Si la determinación de anticuerpos es negativa y no se evidencia otra enfermedad autoinmune, la presencia de cáncer debe igualmente ser descartada en consideración de la posibilidad de un síndrome paraneoplásico, que está presente en 29% de los pacientes ‘seronegativos’, 17% de los pacientes con anticuerpos anti-HMGCR y 8% de los pacientes con anti-SRP33,34. Excepcionalmente la IMNM muestra una evolución lentamente progresiva, asimétrica, con deformidades esqueléticas, escápula alada y atrofia selectiva de algunos grupos musculares, remedado el patrón de una distrofia muscular, la que debe ser excluida35,36. Si bien en estos pacientes no hay una alteración de parámetros indicativos de autoinmunidad, los títulos de anti-SRP o anti-HMGC suelen estar muy elevados35–37.
Miopatía de sobreposición (OM) y Síndrome Antisintetasa (ASS)De forma similar a lo que ocurre con otras enfermedades autoinmunes del tejido conectivo, el síndrome de sobreposición sobreviene cuando la MII se presenta con signos y síntomas extramusculares como los que se observan en el lupus eritematoso sistémico (LES), esclerodermia o en la enfermedad mixta del tejido conectivo. Estas manifestaciones son por ejemplo artritis, enfermedad pulmonar intersticial, fenómeno de Reynaud o síndrome de sicca32. El síndrome antisintetasa (ASS) es la forma más común de miositis de sobreposición (OM). Es una forma particularmente severa de MII definida por la presencia de anticuerpos antisintetasa (más comúnmente anti Jo-1; ver Tabla 2), asociados enfermedad pulmonar intersticial (ILD), síndrome de Reynaud, fiebre, poliartritis distal simétrica de manos y pies, erupciones cutáneas (“rash”) pudiendo ser indistinguibles a los de la DM, así como las “manos de mecánico”, consistentes en lesiones hiperqueratósicas escamosas, simétricas, no pruriginosas (pápulas o placas lineales en la cara lateral de los dedos) en particular el I y II dígitos, pudiendo extenderse hasta los pulpejos y que pueden presentarse también en los pies (“pies de mecánico”)4,22,37–39. Recientemente se ha demostrado que las alteraciones histopatológicas de la IMNM por anticuerpos anti-Jo1 tiene un patrón característico, el que puede ser confundido con DM de no estudiarse correctamente41. La enfermedad pulmonar intersticial está presente entre 79% y 95% de los casos y puede preceder a la miositis en hasta el 50% de los pacientes25.
Miositis por Cuerpos de InclusiónLa miositis por cuerpos de inclusión o también conocida como la forma esporádica de IBM (sIBM), es la forma más común de miopatía inflamatoria en personas mayores de 50 años12,13. Debe distinguirse de las miopatías por cuerpos de inclusión hereditarias (HIBM), que corresponden a un grupo diferente de miopatías genéticamente determinadas por mutaciones en diferentes genes, con las que la sIBM comparte algunas características histopatológicas en común, pero sin inflamación13. En la miositis por cuerpos de inclusión, el cuadro comienza de manera insidiosa, generalmente de forma asimétrica, progresando durante años, lo que a veces hace difícil distinguirlo de una enfermedad de motoneurona o una distrofia de presentación tardía3. Los hallazgos característicos de la sIBM son el comienzo distal en los flexores de los dedos en la extremidad superior, con atrofia del antebrazo (Figura 1c), del compartimiento anterolateral de las piernas (dorsiflexión del pie), asociados con atrofia de los cuádriceps3–5,13. Los pacientes se quejan de dificultad para subir escaleras, pararse desde una silla y caídas frecuentes. A menudo la dificultad mayor es para manipular objetos o hacer fuerza con las manos (Ej. destapar una botella) debido a la debilidad de los flexores largos de los dedos. La debilidad de extremidades suele acompañarse de debilidad facial moderada y de un compromiso de la musculatura axial, que puede resultar en camptocormia o un síndrome de cabeza caída3,42. La dificultad para tragar (disfagia), ocurre en más de la mitad de los casos42. Los pacientes presentan un aumento de la incidencia de polineuropatía. Si bien los hallazgos semiológicos son característicos en sIBM, el diagnóstico se hace a menudo con un promedio de 5 años de retraso, por exclusión en pacientes diagnosticados con PM que no responden al tratamiento inmunosupresor o por una combinación de los hallazgos clínicos, electrodiagnósticos y patológicos3,13. La CK suele estar elevada hasta cinco veces sobre el valor de referencia, pero puede estar normal. Los títulos de anti-cN1A están elevados en un 60-70% de los pacientes con sIBM43, sin embargo pueden también estarlo en la DM (15%), PM (5%), el lupus eritematoso sistémico (14-20%) y en el síndrome de Sjögren (23-36%) por lo que no son específicos43,44. La RM magnética es útil para identificar el patrón de afección característico en la sIBM y permite excluir otras miopatías con RM distintiva13. El estudio electrofisiológico es de difícil interpretación ya que combina hallazgos miopáticos y neurogénicos (o pseudoneurogénicos), o que hace que se confunda con una enfermedad de neurona motora o un compromiso radicular13,45. La electromiografía del flexor profundo de los dedos a diferencia de otros músculos, habitualmente muestra un trazado miopático y ayuda en el diagnóstico46. La biopsia muscular muestra hallazgos que forman parte de los criterios diagnósticos de sIBM. Además de infiltrados inflamatorios consistentes con una polimiositis, se observan vacuolas ribeteadas, depósitos amiloideos generalmente próximos a las vacuolas ribeteadas (cuerpos de inclusión), positivos con Rojo Congo o cristal violeta, fibras rojas rasgadas, así como fibras COXc negativas, causadas por deleciones del ADN mitocondrial y marcadores de degeneración (Ej. p62) que pueden ser identificados por inmunohistoquímica. Si bien estos hallazgos no siempre están presentes o no son específicos de sIBM, su presencia en el contexto clínico adecuado permite el diagnóstico positivo de sIBM o la exclusión de otros diagnósticos. De hecho, se ha demostrado que la combinación del compromiso de los flexores largos de los dedos y de los cuádriceps, con las vacuolas bordeadas o inflamación de fibras no necróticas en la biopsia, son los hallazgos que mejor especificidad y sensibilidad tienen para el diagnóstico de sIBM3,4,13.
ESTRATEGIA DIAGNÓSTICAEl enfrentamiento diagnóstico se basa en una anamnesis y una evaluación clínica rigurosas, en combinación con el uso racional de los métodos complementarios de diagnóstico. Una vez establecida la sospecha de MI, la determinación de los niveles de CK es de utilidad ya que nos permite confirmar la afección muscular y a su vez orientar sobre el tipo de MI presente. Los estudios electrofisiológicos y la RM permiten confirmar la presencia de una miopatía, pero su mayor utilidad es para excluir otros diagnósticos diferenciales, más que para establecer el de una MI, ya que en las miopatías inflamatorias no hay hallazgos específicos con estos métodos de pesquisa. La titulación de MSA y MAA es de suma importancia para determinar el subtipo de MI, así como pesquisar comorbilidad asociada a la MII. La confirmación diagnóstica se logra a través del estudio histopatológico de la biopsia muscular la que es indispensable para tipificar la MI, así como para excluir la posibilidad de otros diagnósticos, especialmente miopatías distróficas (Ej. disferlinopatía, FSHD, LGMD), metabólicas (Pompe, McArdle, lipidosis) y degenerativas (miopatías miofibrilares, HIBM), todas las que se pueden confundir con una MI. De acuerdo con los criterios de la EULAR, la biopsia muscular sólo podría obviarse excepcionalmente, en casos muy típicos de DM juvenil16.
SEROLOGÍAHasta en un 60% de los pacientes con MII los títulos anticuerpos contra complejos moleculares relacionados con el ARN nuclear o contra antígenos citoplasmáticos se hallan elevados3,7. Si bien el rol patogénico de estos anticuerpos no está dilucidado, su determinación es fundamental para el diagnóstico y el pronóstico de las MII. Algunos de estos anticuerpos son específicos (MSA) para ciertas formas fenotípicas de MI; mientras que otros están asociados de forma inespecífica (MAA)7,33. En la Tabla 3 se detallan los MSA y MAA más frecuentes.
Los anticuerpos denominados genéricamente “antisintetasa” por estar dirigidos contra distintas aminoacil-tARN sintetasas, están presentes en 20-30% de los pacientes con MII3,7,38,39. Son un grupo de 8 anticuerpos entre los que destaca el anti-Jo1 que está presente en 75% de los pacientes y está asociado con el síndrome antisintetasa (ver arriba)3,33,38,39. Los anticuerpos MSA relacionados con IMNM están dirigidos más frecuentemente contra la proteína traslacional transportadora SRP37 o contra la 3-hidroximetil-glutaril-coenzima-A-reductasa (HMGCR), que es a su vez el blanco farmacológico de las estatinas40. Este último se observa hasta 22% de los pacientes con IMNM, independientemente de su exposición a estatinas, y su título correlaciona con el nivel de CK y el grado de debilidad7,40. Los anticuerpos asociados a DM incluyen el anti-Mi2 relacionado con las lesiones cutáneas características, el anti-MDA5 en la DM amiopática o la enfermedad pulmonar intersticial, y el anticuerpo anti-TIF1γ y el anti-NPX2 generalmente asociados con la presencia de neoplasia en pacientes adultos con DM3,7,33. Como se mencionó más arriba, el anticuerpo anti–nucleotidasa citosólica 5′1A (anti-cN1A) está elevado en un alto porcentaje de los pacientes con IBM, pero la sensibilidad y especificidad de esta anticuerpo es variable3,43.
BIOPSIA MUSCULARDentro del algoritmo diagnóstico de una MII, la biopsia muscular es de fundamental importancia. El patrón histológico permite establecer el diagnóstico de certeza de la miopatía inflamatoria y excluir otras posibles causas, en particular distrofias musculares30,35,36, además que permite la tipificación de la miopatía inflamatoria2,3,29,31,41. Es importante considerar que distintas formas de distrofia muscular cursan con infiltrados inflamatorios en la biopsia como por ejemplo en la disferlinopatía, la calpainopatía, la distrofia facio-escápulo-humeral, las distrofinopatías; así como también algunas miopatías tóxicas y endocrinológicas2,3,30.
Se debe seleccionar un músculo clínicamente afectado de forma moderada, y que no muestre una gran atrofia. La resonancia magnética puede ser útil en dicha selección, pero no es indispensable. La biopsia debe ser preferentemente a cielo abierto para garantizar un tamaño adecuado de la muestra, en consideración que la afectación inflamatoria del músculo es focalizada y variable. Asimismo debe ser adecuadamente procesada por congelación, ya que los cortes hechos con criostato permiten técnicas histoenzimológicas e inmunohistoquímicas estandarizadas, para la determinación de la expresión del complejo mayor de histocompatibilidad HLA-I, depósitos de complemento y tipificación de poblaciones linfocitarias o la acumulación anormal de moléculas2,41. Es igualmente necesario, para determinar la presencia o ausencia de proteínas estructurales causales de distrofia, relevantes para el diagnóstico diferencial30,35,36. Habitualmente los estudios de microscopía electrónica no son necesarios y sólo son utilizados en casos específicos o con fines de investigación científica2. Cuando los hallazgos de la biopsia son típicos de DM, PM, IMNM o IBM no es mayor problema establecer el diagnóstico3,4. Sin embargo, la interpretación puede verse complicada en aquellos casos en que no hay hallazgos característicos o estos son insuficientes, se hace necesario establecer un correlato clínico acucioso, o repetir la biopsia para su interpretación correcta.
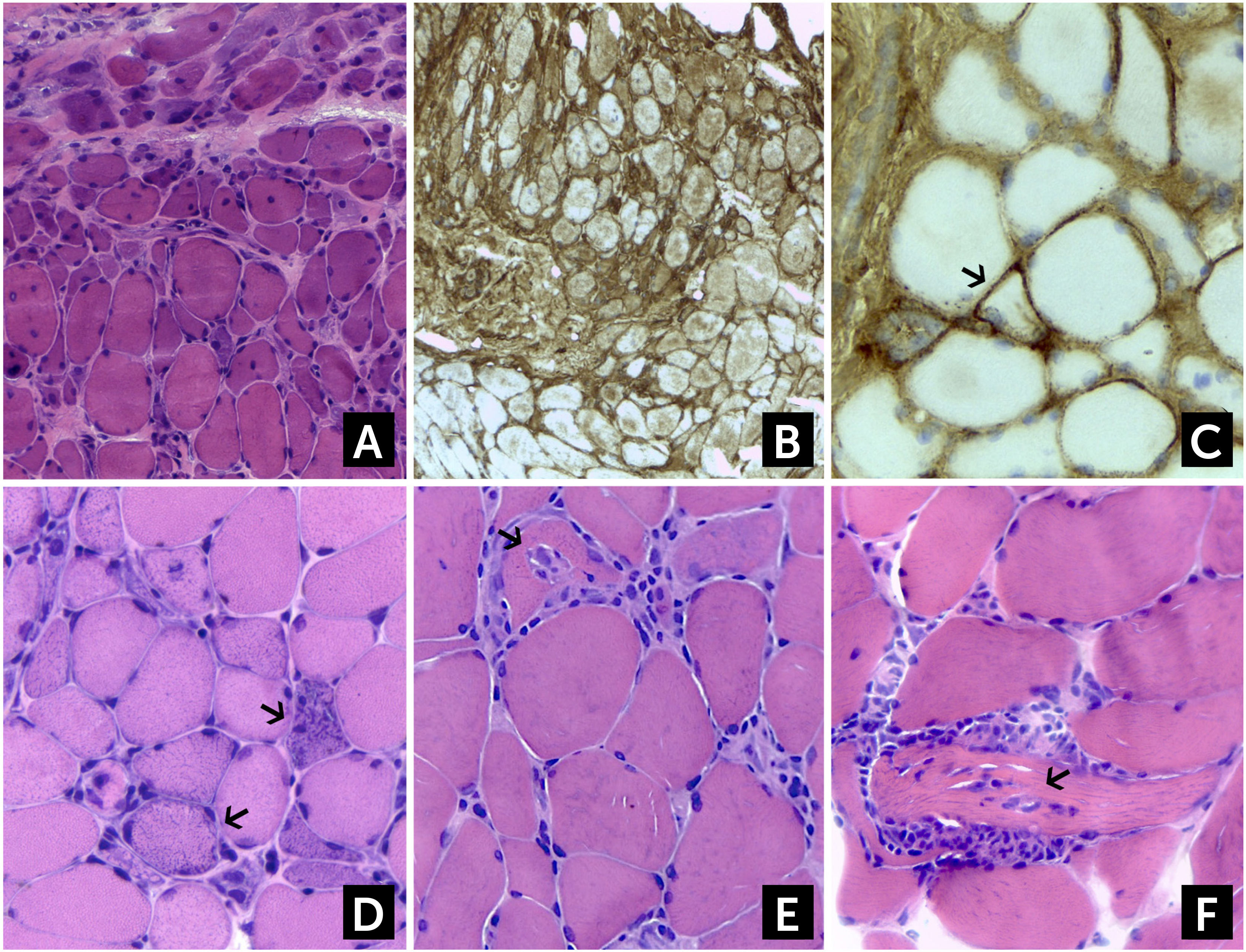
En la DM el infiltrado inflamatorio, predominantemente CD4+, es perivascular o se halla en la periferia del fascículo y se asocia con la típica atrofia perifascicular (Figura 2A). Este último hallazgo es suficiente para sospechar DM aun en ausencia de infiltrados inflamatorios2,3. El número de capilares que rodean las fibras musculares se halla reducido. La inmunohistoquímica revela una sobreexpresión de HLA-I y depósitos del complejo de ataque de membrana (C5b-9) en los capilares (Figura 2B-C).
, Miopatía Necrotizante Inmuno-Mediada (IMNM) y Miositis por Cuerpos de Inclusión (sIBM) (A) Atrofia perifascicular en DM. En el mismo paciente, (B) la biopsia muestra un aumento difuso de la expresión del complejo mayor de histocompatibilidad I (HLA-I), y (C). hay un aumento de depósitos de complemento C5b-9 a nivel de fibras aisladas (flechas), que son característicos de DM. (D) En la IMNM se observan fibras necróticas aisladas, sufriendo miofagocitosis (flechas). Los infiltrados inflamatorios están ausentes o son muy escasos. (E, F) sIBM, los infiltrados inflamatorios intersticiales invaden fibras de aspecto conservado, que a veces producen el efecto de “tunelización” (flechas). (A, D, E y F) hematoxilina-eosina; (B y C) Reacción Inmunohistoquímica para HLA-I y C5b-9 respectivamente.")
Hallazgos histológicos en Dermatomiositis (DM), Miopatía Necrotizante Inmuno-Mediada (IMNM) y Miositis por Cuerpos de Inclusión (sIBM)
(A) Atrofia perifascicular en DM. En el mismo paciente, (B) la biopsia muestra un aumento difuso de la expresión del complejo mayor de histocompatibilidad I (HLA-I), y (C). hay un aumento de depósitos de complemento C5b-9 a nivel de fibras aisladas (flechas), que son característicos de DM. (D) En la IMNM se observan fibras necróticas aisladas, sufriendo miofagocitosis (flechas). Los infiltrados inflamatorios están ausentes o son muy escasos. (E, F) sIBM, los infiltrados inflamatorios intersticiales invaden fibras de aspecto conservado, que a veces producen el efecto de “tunelización” (flechas). (A, D, E y F) hematoxilina-eosina; (B y C) Reacción Inmunohistoquímica para HLA-I y C5b-9 respectivamente.
En la IMNM el hallazgo predominante es la presencia de fibras necróticas aisladas, regularmente distribuidas e invadidas por macrófagos, en ausencia o con muy escasos infiltrados inflamatorios (Figura 2D). La sobreexpresión de HLA-I se halla mayormente limitada en las células necróticas y en algunas fibras de aspecto preservado. Los depósitos de complemento (C5b-9) pueden verse ocasionalmente en la pared de vasos engrosados. Como se comentó anteriormente, en la miopatía necrotizante asociada a anticuerpos anti Jo-1 se ha demostrado un patrón histopatológico característico, consistente en necrosis y depósitos de C5b-9 en áreas perifasciculares, inflamación perimisial y alrededor de los vasos y una sobreexpresión de HLA-I con refuerzo perimisial, el que puede ser confundido con DM de no estudiarse correctamente41. En la IMNM por anticuerpos anti-HMGCR suele haber necrosis aislada de fibras, con escasos infiltrados mononucleares, en particular en fibras necróticas, una marcación para HLA-I aumentada en fibras necróticas y de forma más tenue que en otras IIM en fibras de aspecto normal, y escasa marcación con C5b-9 en aisladas fibras no necróticas10,11.
En la PM y la IBM el tejido muscular muestra infiltrados inflamatorios multifocales, predominantemente T CD8+, que invaden a nivel endomisial y fibras de aspecto aparentemente normal (Figura 2E-F). La expresión de HLA-I se halla uniformemente aumentada, pero no se observan depósitos de complemento (C5b-9). Como se explicó más arriba, en la sIBM, además del patrón descrito para PM, se observan vacuolas ribeteadas, depósitos amiloideos Rojo Congo positivos (cuerpos de inclusión), así como fibras rojas rasgadas con Tricrómico de Gomori modificado y fibras COXc negativas. Finalmente hay un número de proteínas que se acumulan en las fibras en degeneración, susceptibles de detectarse por inmunohistoquímica, pero que no son específicas de IBM ya que se encuentran en otras miopatías neurogénicas y degenerativas2,13,29,44.
RESONANCIA MAGNÉTICAEl estudio imagenológico por RM de los músculos ayuda a confirmar la presencia y magnitud del compromiso muscular, determinar la distribución de este, y contribuir la selección apropiada del músculo para biopsia3,6,13. En los pacientes en los que la MI no está confirmada, la RM puede contribuir a identificar una distrofia muscular o una IBM, ya que estas presentan un compromiso selectivo de grupos musculares determinados. Por ejemplo en IBM se identifica el compromiso de los flexores largos de los dedos en los antebrazos, del cuádriceps en los muslos y a veces de los músculos del compartimiento antero-lateral de la pierna. En las MI en general, las alteraciones son más difusas y multifocales y no suelen seguir un patrón de distribución particular, como sí observamos en los procesos distróficos y degenerativos. La RM debe incluir cortes axiales de las extremidades y tronco en diferentes niveles, así como secuencias ponderadas en T1, T2 y STIR, lo que permite diferenciar el reemplazo fibroadiposo del edema. Es importante destacar que la presencia de edema en la RM, no siempre indica inflamación, como ocurre por ejemplo en procesos neurogénicos.
ELECTROMIOGRAFÍA Y VELOCIDAD DE CONDUCCIÓN NERVIOSA (EMG Y VCN)En un paciente que se presenta con un cuadro clínico característico de miositis, el estudio electrofisiológico sólo sirve para confirmar la presencia de miopatía, y por lo mismo no es necesario2,3. Sin embargo, puede ser utilizado para excluir otros diagnósticos diferenciales4. La conducción nerviosa suele ser normal, o puede mostrar una discreta caída de amplitud de los potenciales de acción motores en algunos casos de miopatía severa47. Los hallazgos EMG característicos de una miopatía son el aumento de la actividad de inserción y de la actividad espontánea con fibrilaciones, ondas agudas positivas, y ocasionalmente descargas pseudomiotónicas y/o descargas repetitivas complejas3,4,47. La presencia de esta última forma de onda indica cronicidad, que el cuadro ha estado presente por varios meses y puede ayudar a dar una idea de la temporalidad. Los potenciales de acción son polifásicos, de baja amplitud y reclutamiento precoz. Los músculos paraespinales son los más afectados y algunos autores sugieren que deben explorarse de forma rutinaria47,48. Dado que estos hallazgos son inespecíficos y pueden estar presentes en miopatías metabólicas y distróficas, su interpretación debe hacerse cautelosamente en consideración del correlato clínico e histopatológico del caso.
TRATAMIENTOEl tratamiento de las MI tiene tres pilares fundamentales: la identificación y erradicación de factores o agentes causales, el tratamiento farmacológico a partir de agentes inmunomoduladores y/o inmunosupresores, y finalmente la terapia física. Estas tres intervenciones no son necesariamente excluyentes y pueden superponerse dependiendo del caso. Por ejemplo, en una miositis infecciosa, el tratamiento adecuado de la infección permitirá la remisión del cuadro; frente a un síndrome paraneoplásico, el tratamiento de tumor primario es mandatorio. En las miopatías gatilladas por agentes miotóxicos, como por ejemplo las estatinas, estas deberán suspenderse. Si la miopatía es causada por el agente, la suspensión del mismo inducirá una remisión de la MI en 4-6 semanas. Si el cuadro persiste, es indicativo que hay un fenómeno autoinmunológico subyacente y debe ser tratado con agentes inmunomoduladores y/o inmunosupresores3,23. El abordaje farmacológico tiene por objetivos primero, inducir una remisión de la MI y segundo, mantenerla23. La inducción de la remisión se realiza habitualmente con metilprednisolona en bolos endovenosos seguidos de prednisona por vía oral, o con prednisona oral desde el inicio, dependiendo de la gravedad del cuadro. El tratamiento debe mantenerse hasta que haya una mejora objetiva de la fuerza muscular de acuerdo a las escalas de valoración funcional, lo que generalmente va acompañado de descenso significativo de los niveles de CK (hasta dos veces por sobre el valor normal)23. Si el uso de corticoides no es suficiente para inducir la remisión, el uso de gamaglobulina endovenosa, ciclofosfamida o rituximab debe ser considerado. La mantención de la reemisión se lleva a cabo a través de la introducción agentes inmunosupresores ahorradores de esteroides como azatioprina, micofenolato o metotrexato siendo este último el que ha mostrado mayor efectividad en las MI44.50. El uso de gamaglobulina endovenosa ha demostrado particular eficacia en el tratamiento de la IMNM causada por HMGCR, así como para disminuir la disfagia en la IBM3,13,44,50. Este abordaje es efectivo en la mayor parte de los pacientes. En los casos refractarios, se ha utilizado agentes inmunosupresores más potentes como la ciclosporina o la ciclofosfamida, con efectividad variable3, o bien una combinación de inmunosupresores23. Se debe tener en cuenta que los esquemas terapéuticos actualmente aceptados están mayormente basados en estudios observacionales, ya que debido en parte a la baja frecuencia de las MII, no ha sido posible obtener resultados concluyentes de muchos ensayos clínicos controlados50. Los anticuerpos monoclonales dirigidos contra subpoblaciones linfocitarias como el rituximab han demostrado efectividad en u algunos casos, aunque la evidencia para su uso está basada en reportes de casos o en pequeñas series de pacientes. Un estudio randomizado con rituxmab en MI refractaria mostró mejoría en un 83% de los casos, pero no alcanzó el objetivo primario, probablemente debido a fallas metodológicas por el cruce temprano entre placebo y rituximab, no dando suficiente tiempo para registrar diferencias49. El uso experimental de alemtuzumab y terapia génica con folistatina han demostrado resultados clínicos alentadores en IBM50.
En tercer lugar, el abordaje fisiátrico de las MI desde un comienzo, basado en ejercicios de resistencia, no extenuantes y un manejo integral según las necesidades del paciente, debe ser implementado tempranamente, de forma gradual, acompañando la mejoría sintomática determinada por la terapia inmunomoduladora.
Declaración de interésLos autores declaran no tener conflictos de interés.
FinanciamientoProyecto FONDECYT 1151383 a JB.