



PROGRESOS DE OBSTETRICIA Y GINECOLOGÍA
Volumen 41 Número 8 Octubre 1998
Casos clínicos
Síndrome de feminización testicular
Testicular femininization syndrome
M.ª J. Pablos
A. Hernández
N. López
Departamento de Obstetricia y Ginecología
Servicio de Ginecología Oncológica
Hospital General Universitario Gregorio Marañón. Madrid
Correspondencia:
M.ª Jesús Pablos Antona
José Anduiza, 13, 4.º D
28034 Madrid
Aceptado para publicación 13/7/98
Pablos MªJ, Hernández A, López N. Síndrome de feminización testicular. Prog Obstet Ginecol 1998;41:489-492.
INTRODUCCIÓN
El síndrome de feminización testicular es la forma más común de pseudohermafroditismo masculino. Su frecuencia es de uno de cada 20.000-64.000 varones. Este cuadro resulta de la insensibilidad del órgano diana a los andrógenos por una alteración en el receptor; como consecuencia estos pacientes presentan un fenotipo femenino con vagina ciega, ausencia de genitales femeninos internos y un cariotipo XY.
Presentamos el caso de una mujer con amenorrea primaria que de forma casual se le detectó una masa pélvica. Durante su estudio se demostró la ausencia de genitales internos y un cariotipo XY. Se realizaron pruebas complementarias y se diagnosticó de síndrome de Morris.
CASO CLINICO
Paciente de 62 años de edad que en una revisión por un angioma hepático se le detecta una masa pélvica. El interrogatorio demuestra que se trata de una paciente con amenorrea primaria que no ha sido estudiada ginecológicamente. Sólo tiene un hermano fallecido por carcinoma pulmonar. Como antecedentes personales presenta un angioma hepático congénito, HTA en tratamiento y una intervención a los siete años de hernia inguinal bilateral.
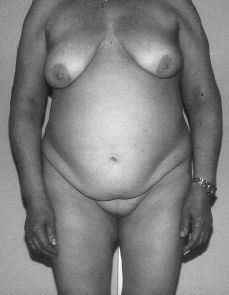
A la exploración, presenta caracteres sexuales secundarios femeninos con desarrollo mamario armónico pero ausencia de vello pubiano (Fig. 1). Los genitales externos eran normales, con una vagina ciega (Fig. 2). Por tacto rectal se palpa en fosa ilíaca izquierda una masa de unos 15 x 15 cm dura, movible y algo dolorosa.
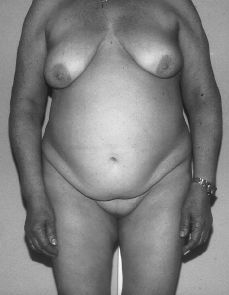
Figura 1.Fenotipo de paciente con síndrome de feminización testicular. Normal desarrollo mamario pero ausencia de vello pubiano.

Figura 2.Genitales externos normales pero con vagina ciega.
Se le practicó una ecografía abdominal que informaba de lesión nodular sólida, hiperecogénica en lóbulo hepático derecho de unos 38 mm de diámetro, muy sugerente de angioma, así como de una imagen quística con tabiques internos de unos 14 cm de diámetro con una imagen cálcica en su interior de unos 2,8 cm. No se logra identificar genitales internos.
Analíticamente presentaba un CA 125 de 59,5 U/ml (elevado), una FSH de 34 mU/ml (normal); LH, 15 mU/ml (normal); testosterona, 0,45 ng/ml (valor de referencia entre 0,1-0,8); estradiol, 10 pg/ml (normal); progesterona, 0,92 ng/ml (normal); androstendiona, 1,7 ng/ml (normal); DHEA-S, 188 microgr/dl (normal). Se le practicó un estudio citogenético de sangre periférica que fue XY, así como una biopsia de piel de labio mayor izquierdo en el que no se reconocían receptores hormonales para andrógenos, estrógenos y progesterona.
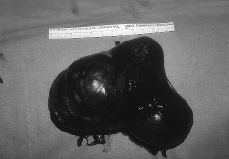
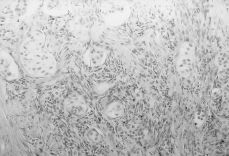
A esta paciente se le practicó una laparatomía, apreciándose una ausencia de útero, una cintilla gonadal derecha con una mínima formación quística de 2 cm y una tumoración de unos 12 cm de diámetro quística, trilobulada, en zona retroperitoneal izquierda, bien capsulada que se extirpa sin rotura (Fig. 3). Se realiza asimismo resección de la cintilla gonadal derecha. Ésta medía 5,3 x 3,6 x 2, 6 cm y estaba constituida por células alargadas y fusiformes que impresionan de estroma ovárico entre las que se reconocen células poligonales similares a las células del hilio ovárico y de Leydig testiculares; también se identifican varios túbulos que impresionan como de origen testicular, al estar recubiertos por células similares a las de Sertoli (Fig. 4). La tumoración anexial izquierda correspondía con un cistoadenocarcinoma seroso de bajo grado de agresividad histológica.
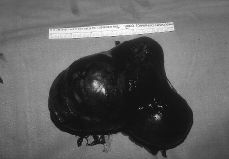
Figura 3. Tumoración anexial izquierda que corresponde a un cistoadenocarcinoma seroso.
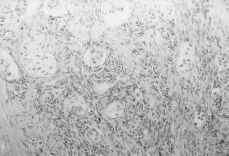
Figura 4. Histología de la cintilla gonadal con células poligonales similares a las células de Leydig y algún túbulo seminífero.
DISCUSION
El síndrome de insensibilidad a los andrógenos fue descrito por Steglehner en 1817. Posteriormente fue Morris quien caracterizó el cuadro y definió los criterios de inclusión, por lo que también es conocido como síndrome de Morris (1). Este cuadro resulta de la insensibilidad del órgano diana a los andrógenos, bien por la ausencia del receptor citoplasmático para los andrógenos o en defectos en su capacidad de fijación o una alteración de los mecanismos que ligan al núcleo de la célula los complejos andrógenos-receptor. Esto produce un fenotipo femenino con cariotipo masculino.
Normalmente la diferenciación sexual masculina depende de la testosterona y del factor inhibidor mülleriano (MIF), ambos secretados por los testículos fetales. La testosterona es directamente responsable de la diferenciación de los conductos wolfianos hacia epidídimo, conducto deferente y vesículas seminales, y es convertida por la 5-alfa-reductasa en la célula diana a dehidrotestosterona (DHT) (2). Por tanto la testosterona no es capaz de estimular el desarrollo wolfiano mientras que el MIF inhibe los conductos müllerianos, con lo cual no se desarrolla aparato genital interno, quedando los testículos en el conducto inguinal favoreciendo el desarrollo de hernias inguinales. La heterogenicidad de la expresión fenotípica va a depender de la ausencia del receptor, de la disminución de la afinidad del receptor o de la disminución en el número de receptores. La forma incompleta del síndrome de feminización testicular se denomina también síndrome de Reinfestein y se caracteriza por fenotipo masculino con ginecomastia, aspecto eunucoide y virilización incompleta en el momento de la pubertad.
El gen para el receptor de andrógenos está localizado en el brazo largo del cromosoma X, en posición Xq 11-12 y está compuesto por ocho exones. Este gen sólo se expresaría en presencia del cromosoma Y, por lo que la enfermedad es transmitida por las mujeres y padecida por los varones. En el síndrome de feminización testicular se han descrito un gran número de mutaciones en este receptor (4). La mayoría afectan a un cambio en un nucleótido, lo que produce un nuevo aminoácido en la secuencia de la proteína. Un ejemplo es la mutación descubierta en el exón 4 responsable de un cambio de leucina a arginina en el codon 707 (3). También se ha publicado la delección en el nucleótido 1893 en el exón 2 que cambia el sentido del codon 622 de una cisteína a una señal de finalización (5).
El síndrome de feminización testicular es la forma más común de pseudohermafroditismo masculino y lo encontramos en uno de cada 20.000-64.000 varones nacidos. El cariotipo de estos pacientes es XY, aunque sus testículos están en el abdomen o en el canal inguinal. Los genitales externos tienen una fusión parcial de los labios y un grado variable de clitoromegalia. La vagina es corta y ciega. Con el desarrollo puberal se observa una telarquia normal, pero el vello pubiano y axilar es muy escaso o ausente y no se produce menarquia. Todas las estructuras müllerianas están ausentes y aunque los conductos wolfianos están presentes son generalmente anormales.
Analíticamente, las cifras son variables, pero se encuentran valores aumentados de testosterona, incluso con respecto a un varón normal y aumento de estrógenos. La FSH puede estar normal o aumentada y la LH ligeramente elevada.
Las pacientes consultan al ginecólogo por amenorrea, masa inguinal o ausencia del vello pubiano. La laparotomía ha sido tradicionalmente el método de diagnóstico y tratamiento en estas pacientes, aunque recientemente ha sido publicada la aplicación de la laparoscopia en estos casos (2).
La frecuencia de neoplasia está aumentada (2-5%), especialmente gonadoblastomas y disgerminomas, por lo que la mayoría de los autores defienden la práctica de la gonadectomía a los 16-18 años (1).
BIBLIOGRAFÍA
1 Hakverdi AU, Cüneyt E, Tarner, Meral A y cols. Incomplete androgen insensitivity (testicular feminization) syndrome: two case reports. Acta Obstet Gynecol Scand 1996;75:588-92.
2 Fang-Ping C. Testicular feminization with incomplete Müllerian regression in twin patients: laparoscopic diagnosis and treatment. Acta Obstet Gynecol Scand 1996;75:304-7.
3 Lumbroso S, Lobaccaro JM, Virginie G y cols. A novel substitution (Leu707Arg) in exon 4 of the angrogen receptor gene causes complete androgen resistance. Journal of Clinical Endocrinology and Metabolism 1996;81:1984-8.
4 Denise W, Patterson M, Hughes I. Androgen insensitivity syndrome. Arch Dis Child 1993;68:343-4.
5 Imai A, Lida K, Okano Y y cols. A frame-shift mutation of the androgen receptor gene in a patient with receptor-negative complete testicular feminization: comparison with a single base substitution in a receptor-reduced incomplete form. Amn Clin Biochem 1995;32:482-6.
6 Belsham DD, Pereira F, Greenberg C y cols. Leu-676-Pro mutation of the AR causes complete androgen insensivity syndrome in a large Hutterite Kindred. Hum Mutat 1995;5:28-33.
7 Brown TR. Human androgen insensitivity syndrome. J Androl 1995;16:299-303.
8 Barón J. Classical and incomplete androgen insensitivity syndromes. Ginekol Pol 1994;65:377-86.
9 Collins GM, Kim D, Logrono R y cols. Pure seminoma arising in androgen insensitivity syndrome (testicular feminization syndrome): a case report and review of the literature. Mod Pathol 1993;6:89-93.
10 Sultán C, Lumbroso S, Poujol N y cols. Mutation of androgen receptor gene in androgen insensitivity syndromes. J Steriod Biochem Mol Biol 1993;46:519-30.
11 Chiba W, Matsubara Y, Sawai S y cols. A case of neurinoma combined with testicular feminization syndrome. Nippon Kyobu Shikkan Gakkai Zasshi 1993;31:94-8.