



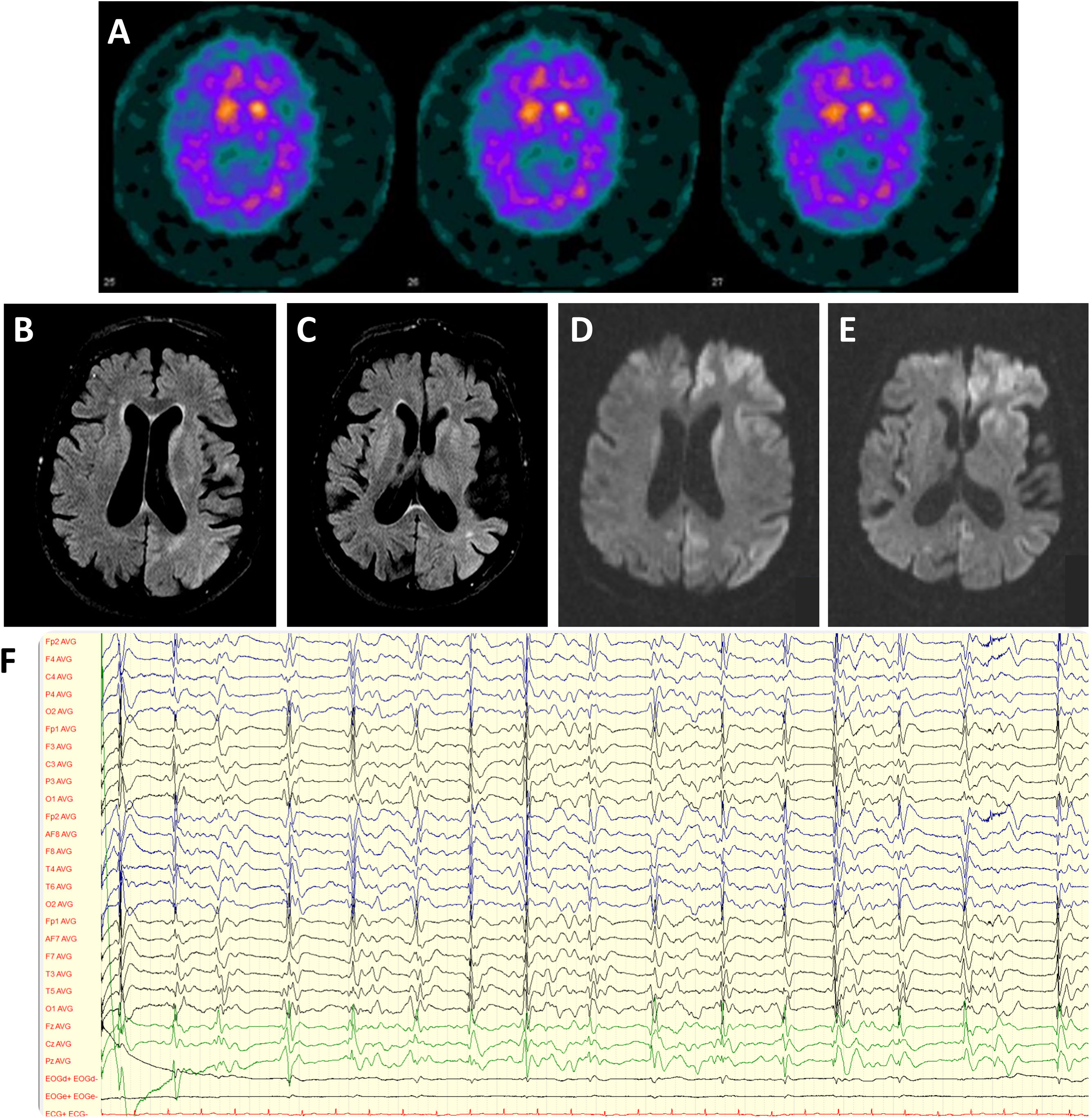
We present the case of a 45 years-old-female who progressively developed bradykinesia and bilateral slightly asymmetrical resting tremor, leading to the diagnosis of PD. There was no family history of parkinsonism, psychiatric disease, or cognitive impairment. 123I-2β-carbomethoxy-3β-(4iodophenyl)-N-(3-fluoropropyl)nortropane (FP-CIT) dopamine transporter SPECT (DaTSCAN) revealed bilaterally decreased dopamine transporter availability in the putamen (Fig. 1A) and the genetic testing of the GBA gene disclosed an heterozygous mutation in exon 9 (p.Asn409Ser). Levodopa replacement therapy was introduced with moderate efficacy. Neuropsychiatric symptoms, including depressed mood, obsessiveness, and psychosis (delusional jealousy) developed shortly after the beginning of the motor symptoms. The patient remained stable for almost 2 decades until 2019, when a progressive worsening of motor symptoms (resting tremor, postural instability and gait impairment) was observed, together with an increase of depressive mood symptoms. One year later, the patient was admitted in the emergency department due to rapidly progressive deterioration of her clinical condition within the last two weeks associated with apathy, loss of speech and walking ability and total dependence for activities of daily living. On neurological examination the patient was awake, directing the eyes to sound stimulus but was unable to speak or follow simple commands. Frequent generalized myoclonus predominantly on the right hemibody and marked startle with auditory stimulus were evident, as well as bilateral symmetric rigidity and bradykinesia. Blood and cerebrospinal fluid (CSF) laboratory studies excluded metabolic imbalances or systemic and central nervous system infections. Brain MRI showed diffuse cortical atrophy predominantly in the temporal lobes and T2 hyperintensity in basal ganglia and parieto-temporal transition; diffusion-weighted imaging (DWI) documented diffusion restriction in caudate and lenticular nucleus, as well as antero-medial frontal, insular and temporo-parietal cortex (Fig. 1B–E). Electroencephalogram revealed generalized periodic discharges at 0.5–1.5Hz with triphasic waves (Fig. 1F). CSF study (Table 1) showed significantly increased in tau protein and positive 14-3-3 protein. Real-time quaking-induced conversion (RT-QuIC) assay confirmed the presence of the pathological form of prion protein in the CSF. During the next month, the patient's neurological status deteriorated with the development of akinetic mutism. She died 10 months after the beginning of the symptoms.
 DaTSCAN revealing bilateral decreased of dopamine transporter availability in the putamen. (B, C) Brain MRI showing diffuse cortical atrophy predominantly in the temporal lobes and hyperintensity in basal ganglia and parieto-temporal transition on Fluid attenuated inversion recovery (FLAIR). (D, E) Diffusion restriction in caudate and lenticular nucleus, as well as antero-medial frontal, insular and temporo-parietal cortex on DWI (diffusion-weighted magnetic resonance imaging). (F) Electroencephalogram revealing generalized periodic discharges at 0.5–1.5Hz with triphasic waves.")
(A) DaTSCAN revealing bilateral decreased of dopamine transporter availability in the putamen. (B, C) Brain MRI showing diffuse cortical atrophy predominantly in the temporal lobes and hyperintensity in basal ganglia and parieto-temporal transition on Fluid attenuated inversion recovery (FLAIR). (D, E) Diffusion restriction in caudate and lenticular nucleus, as well as antero-medial frontal, insular and temporo-parietal cortex on DWI (diffusion-weighted magnetic resonance imaging). (F) Electroencephalogram revealing generalized periodic discharges at 0.5–1.5Hz with triphasic waves.
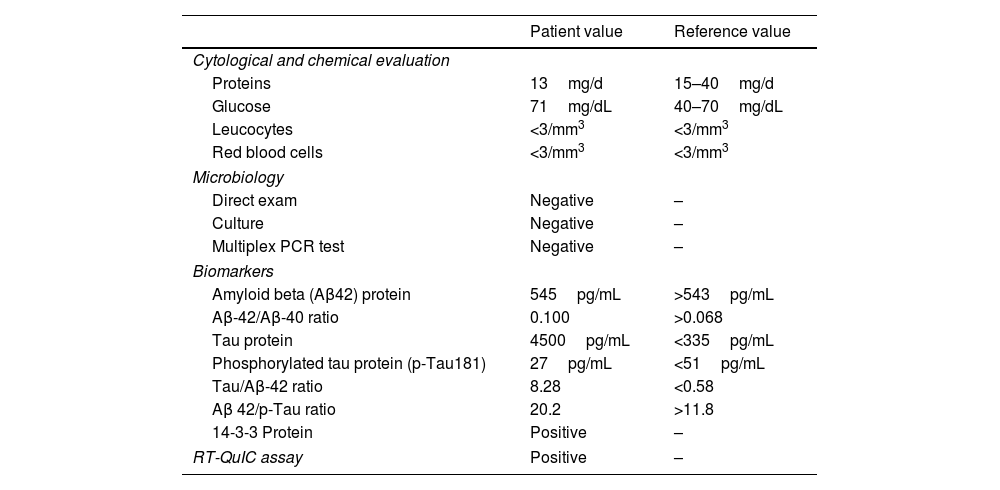
CSF analysis.
| Patient value | Reference value | |
|---|---|---|
| Cytological and chemical evaluation | ||
| Proteins | 13mg/d | 15–40mg/d |
| Glucose | 71mg/dL | 40–70mg/dL |
| Leucocytes | <3/mm3 | <3/mm3 |
| Red blood cells | <3/mm3 | <3/mm3 |
| Microbiology | ||
| Direct exam | Negative | – |
| Culture | Negative | – |
| Multiplex PCR test | Negative | – |
| Biomarkers | ||
| Amyloid beta (Aβ42) protein | 545pg/mL | >543pg/mL |
| Aβ-42/Aβ-40 ratio | 0.100 | >0.068 |
| Tau protein | 4500pg/mL | <335pg/mL |
| Phosphorylated tau protein (p-Tau181) | 27pg/mL | <51pg/mL |
| Tau/Aβ-42 ratio | 8.28 | <0.58 |
| Aβ 42/p-Tau ratio | 20.2 | >11.8 |
| 14-3-3 Protein | Positive | – |
| RT-QuIC assay | Positive | – |
CSF Aβ42, Aβ40, t-Tau and p-Tau181 were determined in the fully automated hemiluminescence enzyme immunoassay platform LUMIPULSE G600II (Fujirebio, Ghent, Belgium), using the Lumipulse G β-Amyloid 1-42, β-Amyloid 1–40, Total Tau and pTau 181 assays, following the manufacturer's instructions.
The GBA gene encodes the lysosomal enzyme glucocerebrosidase (GCase), an enzyme responsible for the final step of degradation of glycosphingolipids. Homozygous GBA mutations are associated with Gaucher Disease, a lysosomal storage disease presenting with systemic and neurological manifestations. Heterozygous GBA mutations are a major genetic risk factor for Parkinson's disease (PD).1 GCase acts as a regulator of α-synuclein homeostasis through its action on protein trafficking, degradation of defective α-synuclein and interaction with membrane lipids.2,3 The onset of GBA-PD is generally 5 years earlier than sporadic PD, with motor symptoms being clinically non-distinguishable from sporadic PD and characterized by early motor complications.4,5 Non-motor symptoms tend to be more prevalent and severe in GBA-PD compared to non-carriers, including early cognitive impairment with predominant affection of executive and visuospatial functions, anxiety, depression, psychosis and sleep disturbances.1,6,7
The presence of rapidly progressive dementia, myoclonus and akinetic mutism associated with typical EEG findings, 14-3-3 protein positivity and positive RT-QuIC assay points towards the diagnosis of probable Creutzfeldt–Jakob disease (CJD) according to the CDC's Diagnostic Criteria 2018.8
The association of parkinsonism with psychiatric symptoms and cognitive dysfunction raises an extensive list of possible diagnoses including Parkinson's disease dementia, iatrogenic parkinsonism, atypical parkinsonism and CJD, which may present itself with parkinsonism.9 In this case, the careful examination of the time of onset and progression of the different symptoms allowed the differential diagnosis between PD dementia and an additional cause of cognitive impairment.
The incidence of CJD in patients with PD is quite low. There are few reports in the literature of CJD in patients with presumed idiopathic PD, as genetic testing was not performed.10–12 To our best knowledge this is the first report of CJD in a GBA-PD patient. Given the low incidence of CJD and the relatively high incidence of PD and heterozygous GBA mutation, both diseases may coexist independently in the same patient. However, the increased susceptibility to neuronal degeneration in GBA mutations carriers has been object of scientific interest and the involved mechanisms remain uncertain. One hypothesis postulate that α-synuclein aggregates might have a prion-like behaviour of cell-to-cell transmission with dopaminergic neuron degeneration.13 However, this hypothesis is revoked in our patient by the presence of positive RT-QuIC assay.
Therefore, we can only speculate the possible impact of heterozygous GBA mutation with consequent impairment of the autophagic-lysosomal pathway including degradation of defective proteins and dysfunction in protein trafficking in the development and clinical evolution of CJD.
Informed consentInformed consent was obtained from the patient for publication of this case report.
Conflict of interestThe authors declare no conflicts of interest and no disclosures relevant to the manuscript.
None.

