



La encefalopatía hiperamonémica inducida por valproato (EHV) es una entidad poco frecuente que se caracteriza clínicamente por un cuadro agudo o subagudo que puede presentar alteración del nivel de consciencia, confusión, signos y síntomas neurológicos focales, y progresar asociando crisis epilépticas, ataxia, estupor y coma1. En la mayoría de los pacientes los niveles de amonio están elevados sin signos de fallo hepático, y también pueden observarse alteraciones en el electroencefalograma (EEG), habitualmente un enlentecimiento delta generalizado1. A pesar de que las crisis epilépticas son uno de los síntomas reconocidos en esta entidad, solo se han comunicado 2 casos de estatus epiléptico inducido por valproato (VPA)2,3. A continuación describimos el caso de una paciente que sufrió un estatus epiléptico no convulsivo (EENC) secundario a una EHV.
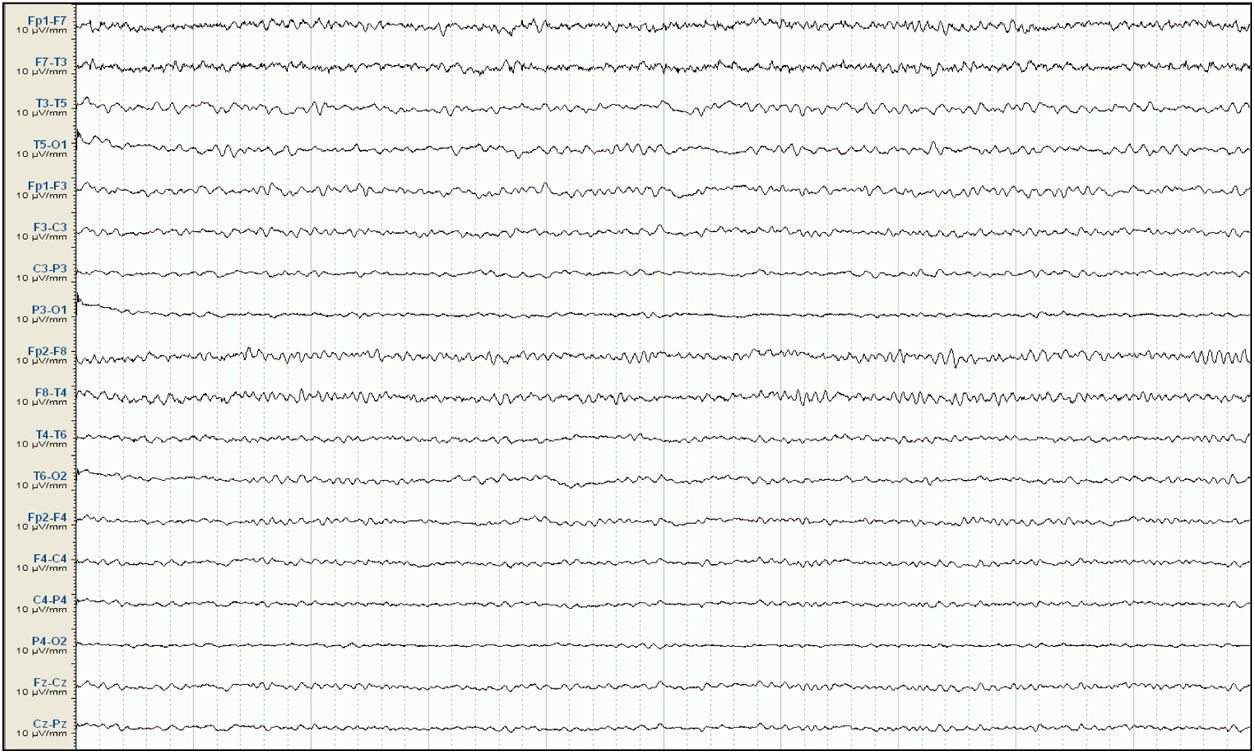
Mujer de 57 años, con antecedentes de enfermedad renal crónica secundaria a nefropatía intersticial, en tratamiento con hemodiálisis desde 2007, con trasplante renal en 2009 y rechazo crónico del injerto, en tratamiento con prednisona 5mg/24h. Seis meses antes del ingreso presenta un primer episodio de crisis tónico-clónica generalizada (CTCG) tras una sesión de diálisis, valorada en urgencias con un estudio analítico general y una tomografía axial computarizada (TAC) cerebral que no mostraron alteraciones significativas, sospechándose como origen un posible desequilibrio metabólico en el contexto de la diálisis. Ante un segundo episodio un mes después, dado que de nuevo no se observaron alteraciones metabólicas significativas, y al observarse en el EEG descargas epileptiformes temporales derechas, se inicia tratamiento con VPA a dosis de 500mg/12h y se remite a consulta externa de neurología para completar estudio y seguimiento. Ante la recurrencia posterior de las crisis, es atendida de nuevo en urgencias y se añade al tratamiento levetiracetam (LEV) 500mg/12h. Aproximadamente un mes antes de su ingreso desarrolla un cuadro de agitación, heteroagresividad e ideas autolíticas, motivo por el cual ingresa en planta de psiquiatría, añadiéndose al tratamiento olanzapina y lorazepam, y disminuyéndose la dosis de LEV. Tras una semana de ingreso se observa fluctuación del nivel de alerta y CTCG de repetición sin recuperación completa del nivel de consciencia, por lo que ingresa en la UCI. En la exploración en ese momento se observa un índice de Glasgow 3, pupilas medias isocóricas y normorreactivas y episodios fluctuantes de movimientos clónicos irregulares de extremidades multifocales, por lo que se procede a intubación orotraqueal y se inicia tratamiento antiepiléptico intravenoso con VPA y LEV, yugulando la actividad motora; en este momento se suspende el tratamiento psiquiátrico. Se realiza un EEG en el que se observa un trazado compatible con EENC (fig. 1), por lo que se asocia lacosamida al tratamiento y se realizan estudios analíticos generales y neuroimagen mediante TAC cerebral sin observarse alteraciones significativas, manteniendo su función renal habitual sin cambios. Se realizan EEG de control sin cambios significativos, por lo que progresivamente se asocia perampanel al tratamiento y se van aumentando las dosis de midazolam y propofol, iniciados en el momento de la intubación. Ante la persistencia del trazado, se amplía el estudio con análisis de LCR, sin alteraciones en bioquímica, citología, PCR de virus neurotropos, anticuerpos antineuronales y negatividad de bandas oligoclonales, así como del estudio de autoinmunidad en suero. En el EEG de control se observa una progresión a un trazado de supresión con brotes en los que se continúan observando grafoelementos agudos mantenidos. En controles analíticos posteriores se observan unos niveles de VPA por debajo de rango terapéutico de forma mantenida, y unos niveles de amonio en suero elevados (65μmol/l, rango de referencia: 9-35), parámetro no valorado previamente, por lo que se decide suspender el tratamiento con VPA 8 días después del ingreso en la UCI. Tras ello, se observa una mejoría progresiva de los controles de EEG con menor actividad aguda, no mantenida, por lo que se va disminuyendo sedación, observándose un trazado prácticamente normalizado a los 7 días (fig. 2), con disminución progresiva de las cifras de amonio hasta su normalización, y mejoría del nivel de consciencia hasta la recuperación completa, sin presentar nuevos episodios epilépticos.

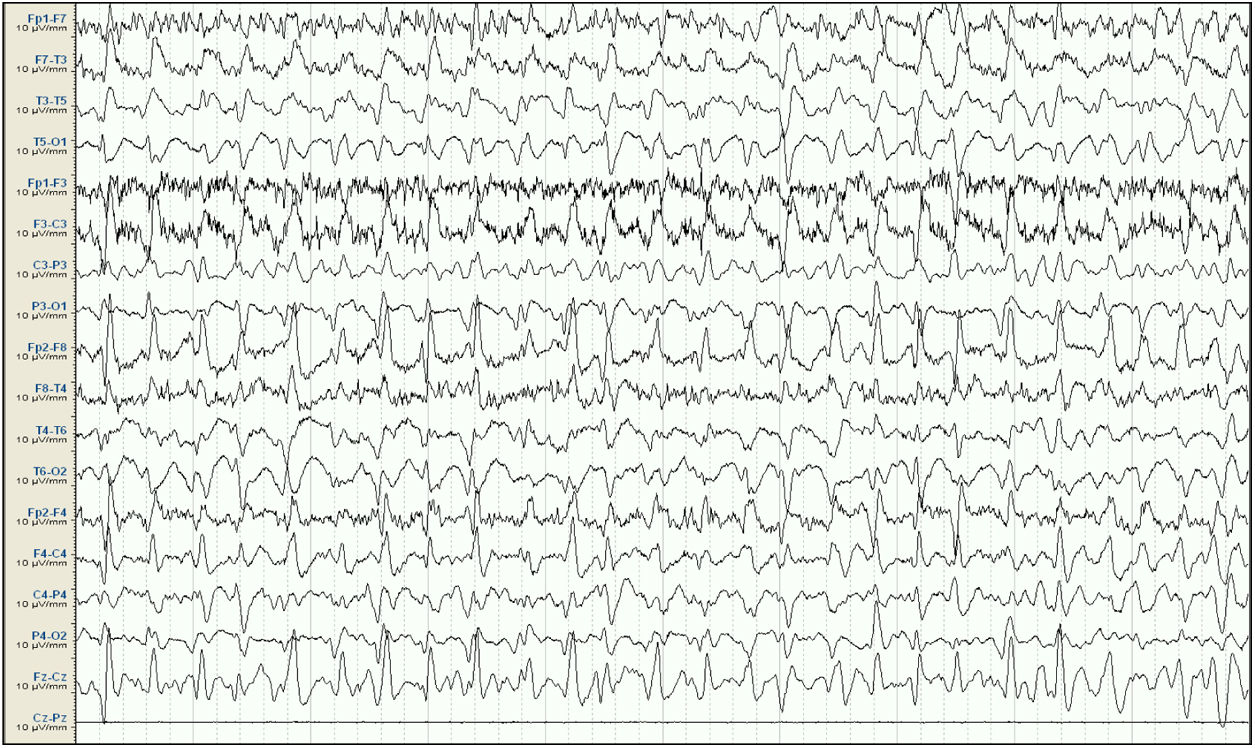
EEG inicial. Sistema 10/20, montaje longitudinal. Actividad de fondo mal diferenciada que es sustituida por descargas epileptiformes difusas de predominio en áreas fronto-centrales derechas, persistentes a lo largo de todo el registro, compatible con estatus epiléptico. Ausencia de señal en la derivación Cz-Pz debido a problemas técnicos.

La EHV es una complicación no habitual del tratamiento con dicho fármaco. Se produce por una interferencia del VPA en el ciclo de la urea inhibiendo la enzima carbamilfosfato sintetasa I (CPS). El VPA se metaboliza extensamente a nivel hepático por 3 vías; en situaciones de altas dosis de VPA o en tratamientos de larga evolución hay una mayor producción de metabolitos como el ácido propiónico o el 4-en-VPA, que producen una reducción de N-acetilglutamato, cofactor que activa la CPS, dando lugar a una inhibición de dicha enzima, interfiriendo por lo tanto en el ciclo de la urea y produciendo hiperamonemia4. Aunque el mecanismo exacto de producción de encefalopatía no es totalmente conocido, se sabe que este exceso de amonio pasa la barrera hemato-encefálica e inhibe el consumo intracelular de glutamato; el incremento de la actividad del receptor NMDA debida al aumento de glutamato extracelular produce, en última instancia, excitotoxicidad, conduciendo finalmente al cuadro de encefalopatía4,5. Otras hipótesis de la fisiopatología del cuadro argumentan que el aumento de producción de glutamina en los astrocitos debido a la eliminación del exceso de amonio cerebral provoca un incremento de la osmolaridad y desplazamiento intracelular de agua, dando lugar a edema celular y finalmente cerebral4,6.
Se han identificado ciertos factores de riesgo para el desarrollo de la EHV, como la politerapia antiepiléptica, observándose con PB, PHE, TPM o LEV asociados a VPA7–9; otros factores asociados son la deficiencia de carnitina y los trastornos del ciclo de la urea5. Sin embargo, los niveles séricos de VPA elevados no se correlacionan directamente con el desarrollo de la EHV10, como en el caso que nos ocupa, en el que se observan unos niveles de VPA bajos.
El diagnóstico de esta entidad debe centrarse en una clínica de encefalopatía de inicio agudo junto con niveles de amonio elevado en el contexto de tratamiento con VPA. Hay que tener en cuenta que pacientes en tratamiento con VPA, sobre todo si reciben otros fármacos antiepilépticos, pueden presentar una hiperamonemia asintomática11. Sin embargo, en pacientes con un declive cognitivo leve más subagudo o crónico, o manifestaciones psiquiátricas en el contexto del tratamiento con VPA, debe tenerse en mente esta entidad ya que puede desarrollarse una forma de presentación más larvada que dificulta notablemente el diagnóstico, como en el caso presentado. En este contexto, se debe llevar a cabo un diagnóstico diferencial con otras encefalopatías subagudas (metabólicas, tóxicas, infecciosas, autoinmunes…), que en nuestro caso resultó negativo. Además de un origen encefalopático, el EENC como entidad también debe descartarse ante un cuadro de alteración de nivel de consciencia, debiendo primero objetivarse mediante EEG y posteriormente plantear su amplio diagnóstico diferencial etiológico.
Hasta el momento se han publicado 2 casos de EENC inducido por VPA; el primero presenta un paciente con una clínica aguda de bajo nivel de consciencia y mioclonías multifocales cuatro días tras el inicio de tratamiento con VPA, con un EEG compatible con EENC2. El segundo nos describe a una paciente con una epilepsia focal farmacorresistente con mal control de crisis que, tras una titulación rápida de VPA hasta 1.600mg en 5 días, desarrolla un estado confusional fluctuante asociado a episodios de desconexión del entorno, con un EEG también compatible con EENC; en este caso los niveles de amonio no estaban elevados3. Nuestro caso se diferencia, y presenta la dificultad diagnóstica añadida, en que el tratamiento con VPA llevaba meses instaurado a las mismas dosis, no se trataba de un cuadro asociado con el inicio de tratamiento como en los dos casos comentados, ni con un aumento de dosificación, lo que condujo al retraso diagnóstico. La ausencia de datos patológicos en el estudio complementario excepto los niveles elevados de amonio, y la mejoría electroclínica observada tras la suspensión de VPA sin realizar otras maniobras terapéuticas simultáneamente, nos hacen pensar en la EHV como el diagnóstico más probable. Por otra parte, tanto en nuestra paciente como en el primer caso comentado2, los niveles de amonio no estaban excesivamente elevados; se ha observado que no existe correlación entre la severidad clínica del cuadro y niveles más elevados de amonio1.
En conclusión, se presenta un caso poco habitual de EENC secundario a una EHV. A pesar de ser una entidad poco frecuente, debemos tenerla en cuenta en el diagnóstico diferencial del EENC refractario sin etiología evidente en pacientes tratados con VPA.
FinanciaciónEl trabajo no ha sido financiado por ninguna entidad pública ni privada.

