



La mutación del gen SYNGAP1 fue descrita por primera vez en 2009 en pacientes con retraso psicomotor no sindrómico y trastornos del espectro autista. Posteriormente, en 2013 se describió como causa de encefalopatía epiléptica y del desarrollo (EED)1. Este gen está localizado en el cromosoma 6p21.32 y codifica la proteína SYNGAP1 (proteína activadora de la GTPasa sináptica 1), regulando el balance excitador/inhibidor de las neuronas hipocampales, en relación con receptores NMDA (N-metil-D-aspartato) y AMPA (ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico). La mayoría de mutaciones son de novo causando una proteína truncada hipofuncionante1,2. Hasta un 98% de los pacientes padecen epilepsia1,3, con un porcentaje de hasta el 50% de farmacorresistencia en algunas series4, siendo las crisis más frecuentemente descritas las ausencias atípicas, las mioclonías palpebrales, las crisis mioclónicas-atónicas con caída, además de las crisis reflejas, en especial con la comida1.
El cannabidiol es un fármaco aprobado para los síndromes de Lennox-Gastaut (SLG) y de Dravet (SD) en adyuvancia con clobazam, en pacientes mayores de 2 años, en Europa, y para el complejo de esclerosis tuberosa (CET), en EE.UU.5. Actúa reduciendo la hiperexcitabilidad neuronal, además de poseer un efecto ansiolítico y regulador del sueño6. Algunos estudios han demostrado la mejoría a nivel cognitivo y conductual en modelos animales y estudio en humanos7, además de eficacia en otras patologías que asocian crisis epilépticas8.
Presentamos el caso de una paciente diagnosticada de encefalopatía por mutación de SYNGAP1 en nuestro centro, siendo tratada con cannabidiol por farmacorresistencia con mejoría clínica importante.
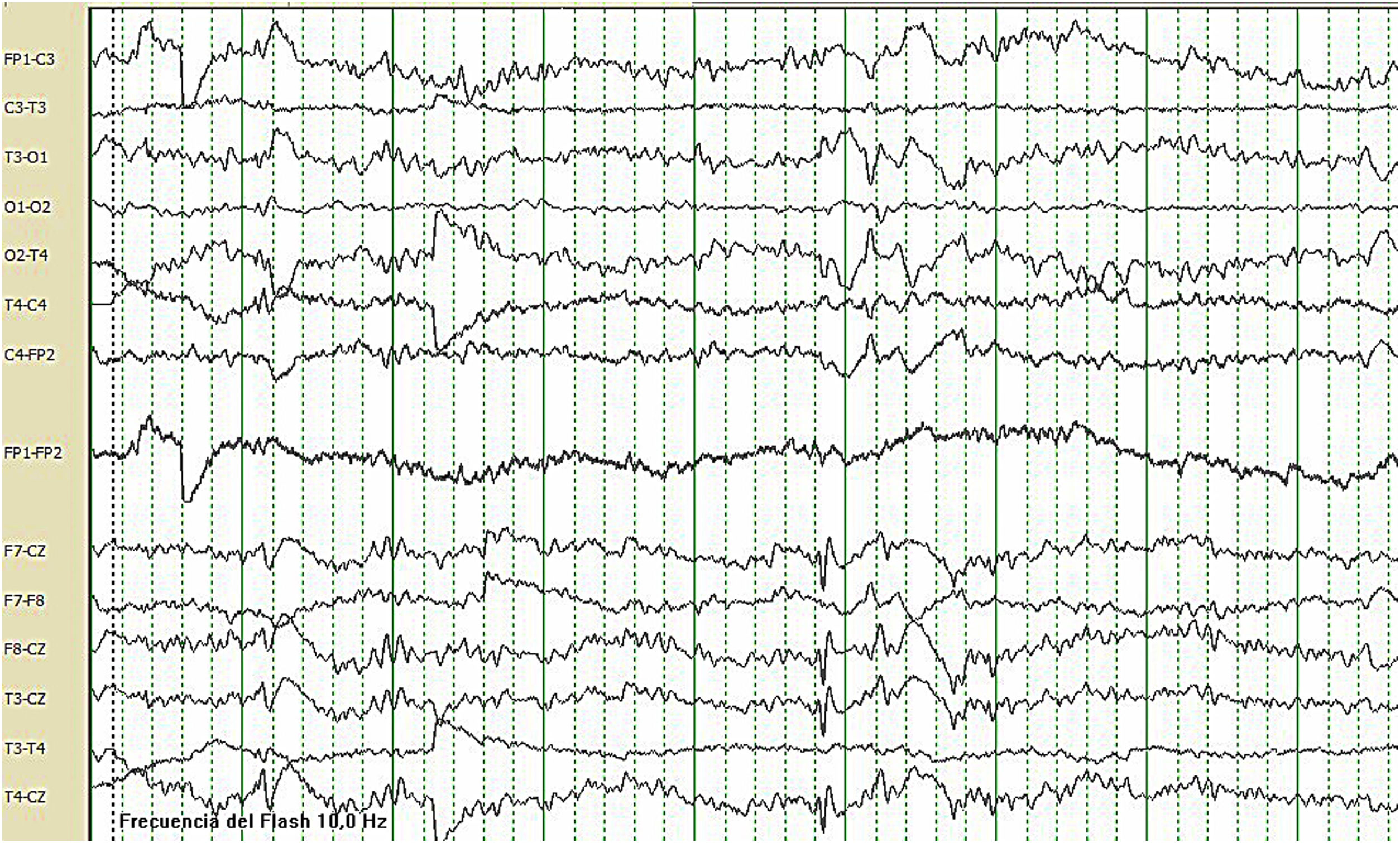
Caso clínicoPaciente mujer de 21 años, sin antecedentes perinatales de interés. Desde los primeros días de vida, comenzó con episodios diarios de desconexión medioambiental, con automatismos orodeglutorios, episodios compatibles con mioclonías palpebrales, axiales y crisis atónicas acompañándose de retraso psicomotor. Más tarde, aparecieron crisis tónico-clónicas generalizadas (CGTC) y crisis gelásticas. Nunca adquirió lenguaje siendo diagnosticada de trastorno del comportamiento del espectro autista, con episodios de alteración de conducta. Se realizó un cariotipo, una resonancia magnética cerebral, estudios genéticos y metabólicos con resultado normal. Al comenzar visitas en la Unidad de Epilepsia de adultos del Hospital Regional Universitario de Málaga (HRUM), la paciente presentaba crisis focales con generalización secundaria y atónicas varias veces al día, además de CGTC dos veces a la semana y mioclónicas frecuentes, habiendo probado numerosas combinaciones de fármacos antiepilépticos (FAE), incluyendo carbamazepina, perampanel, brivaracetam y clonazepam que habían sido retirados por ineficacia, por lo que cumplía criterios de farmacorresistencia. Se encontraba en tratamiento con ácido valproico (900mg al día) y lacosamida (400mg al día). Se realizó una monitorización video-EEG, durante la que no tuvo eventos críticos, pero mostró una actividad de base lentificada compatible con una encefalopatía difusa con superimposición de brotes de ondas delta rítmicas hipervoltadas en regiones centrales bilaterales y vértex (fig. 1). Se realizó un exoma en 2018 detectándose una mutación nonsense de novo en heterocigosis c.1861C>T del gen SYNGAP1, que conlleva el cambio del aminoácido Arg621, truncando la proteína.
 rítmicas hipervoltadas en regiones centrales bilaterales y en vértex.")
Dada la falta de control de las crisis y de la conducta, se decidió iniciar tratamiento con cannabidiol (300mg cada 12h), y clobazam (10mg por la noche) en adyuvancia. A los 3 meses se objetivó la eficacia del nuevo tratamiento en forma de disminución del número de crisis en más de un 50% así como de la duración de las mismas, desapareciendo las crisis atónicas y presentando una mejoría de la eficacia de la medicación de rescate. Asimismo, el mejor control de las crisis ha conllevado una mejoría funcional, recuperando la capacidad de deambulación autónoma y un mejor control conductual.
DiscusiónLa EED por mutación del gen SYNGAP1 es una enfermedad rara recientemente descrita, pero con un síndrome epiléptico característico que combina ausencias con mioclonías palpebrales y crisis mioclónico-atónicas. Hemos presentado el caso de una paciente con EED secundaria a mutación patogénica en el gen SYNGAP1 descrita en el año 20209 que, como peculiaridad, presenta crisis gelásticas. No hemos encontrado ningún caso con este tipo de crisis. Destaca a su vez la buena respuesta a la terapia con cannabidiol. Las mutaciones en este gen inducen un incremento en la proteína del canal de cationes de potencial receptor transitorio miembro 1 de la subfamilia V, uno de los mecanismos que provocan el desequilibrio excitatorio/inhibitorio, y el cannabidiol parece inducir una rápida activación y desensibilización de este receptor8. De hecho, han sido publicadas algunas series de pacientes con buena respuesta clínica1,10. En base a estos datos, decidimos asociar cannabidiol a nuestra paciente como uso compasivo, objetivándose una mejoría clínica, tanto a nivel de control de crisis como a nivel cognitivo y de conducta.
Debido a que existen cada vez más casos descritos en la literatura y la mayor disponibilidad de estudios genéticos, probablemente en los próximos años será posible identificar un mayor número de pacientes, lo que permitirá una mejor caracterización de la enfermedad y obtener datos de eficacia de los tratamientos. El cannabidiol como uso compasivo podría ser un fármaco eficaz en la EED, con necesidad de un mayor número de estudios en el futuro.

