




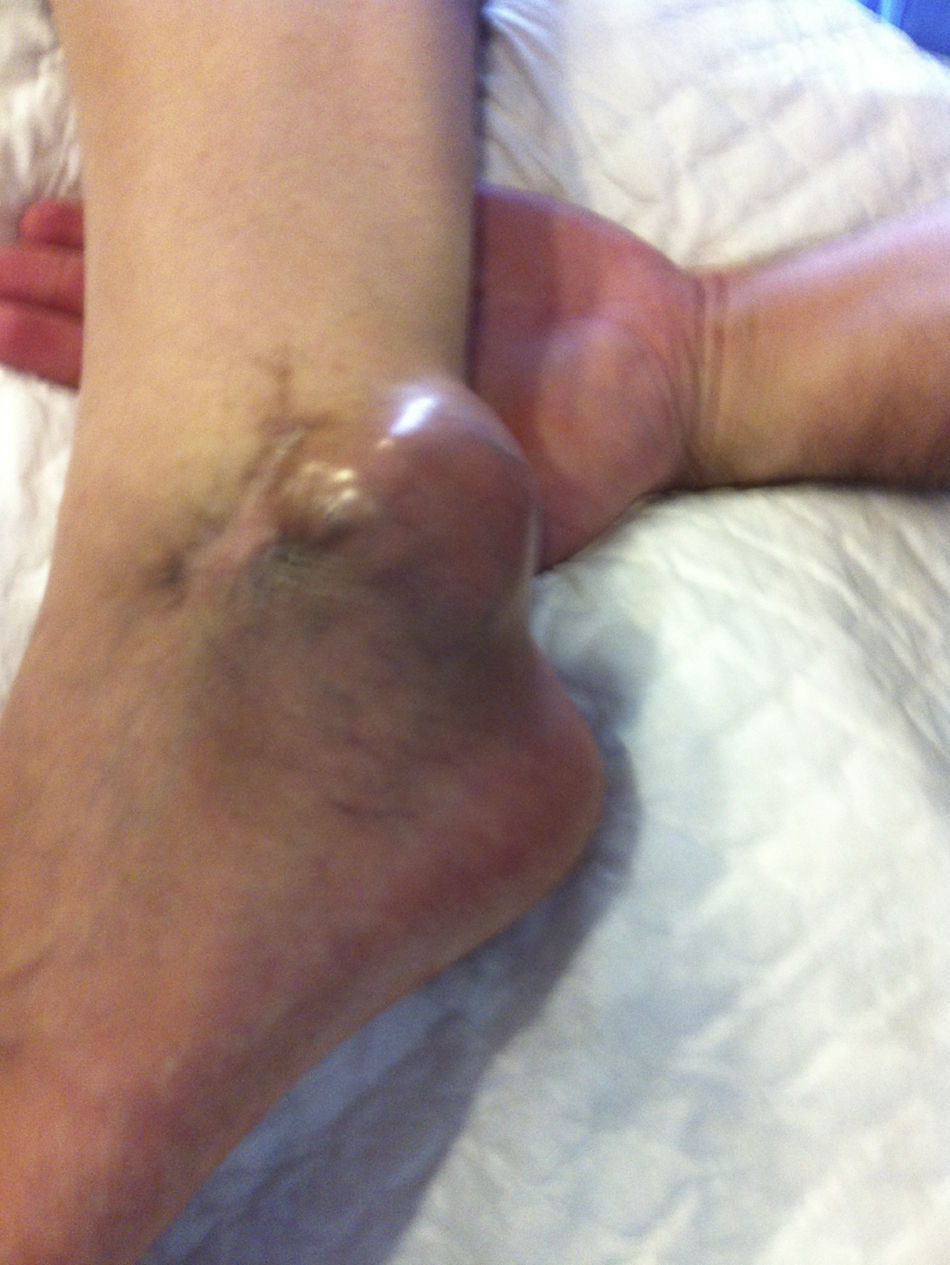
Presentamos el caso de un varón de 37años de edad en seguimiento desde la consulta de atención primaria por tumoración localizada en región retromaleolar del tobillo izquierdo, de consistencia semiblanda, que finalmente se diagnosticó de sarcoma de partes blandas y requirió amputación transtibial izquierda.
El paciente refiere que en agosto de 2013, cuando residía en Guinea Ecuatorial, lugar donde se encontraba trabajando, fue intervenido quirúrgicamente de una tumoración no dolorosa en la región retromaleolar del tobillo izquierdo. Dicha tumoración fue extirpada y no analizada al considerarse que se trataba de un ganglión. A las 48h de la intervención comenzó con fiebre, diagnosticándose de malaria y siendo tratado con Artesunato y Fansidar, presentando inicialmente una buena evolución. Cinco días más tarde la herida comenzó a presentar un exudado purulento, por lo que se pautó tratamiento antibiótico con azitromicina y metronidazol, con escasa respuesta, por lo que se sustituyó por ceftriaxona intravenosa. Ante la evolución tórpida de la herida quirúrgica, el paciente decidió regresar a España, precisando ingreso hospitalario, con diagnóstico de infección de partes blandas por Escherichia coli Blee y Aeromonas hydrophila y dehiscencia de sutura de herida quirúrgica. Al alta, continuó con curas diarias en nuestro centro de salud hasta el cierre completo de la herida en enero de 2014.
Regresa nuevamente a Guinea por motivos de trabajo, pero en marzo de 2014, en la misma zona de la extirpación previa, comienza de nuevo a crecer la tumoración de forma rápida, por lo que el paciente decide finalmente volver a España. Se cita en consulta con su médico de atención primaria, que ante la clínica y los hallazgos exploratorios le remite de forma preferente a cirugía, siendo en mayo del mismo año reintervenido. El examen anatomopatológico de la lesión, tras toma de biopsia, se informa como tumoración mesenquimal maligna. El estudio de extensión con TAC toracoabdominal y gammagrafía ósea fueron normales, y la resonancia nuclear magnética no detectó tumor residual macroscópico, por lo que se decide amputación infracondílea izquierda (fig. 1).

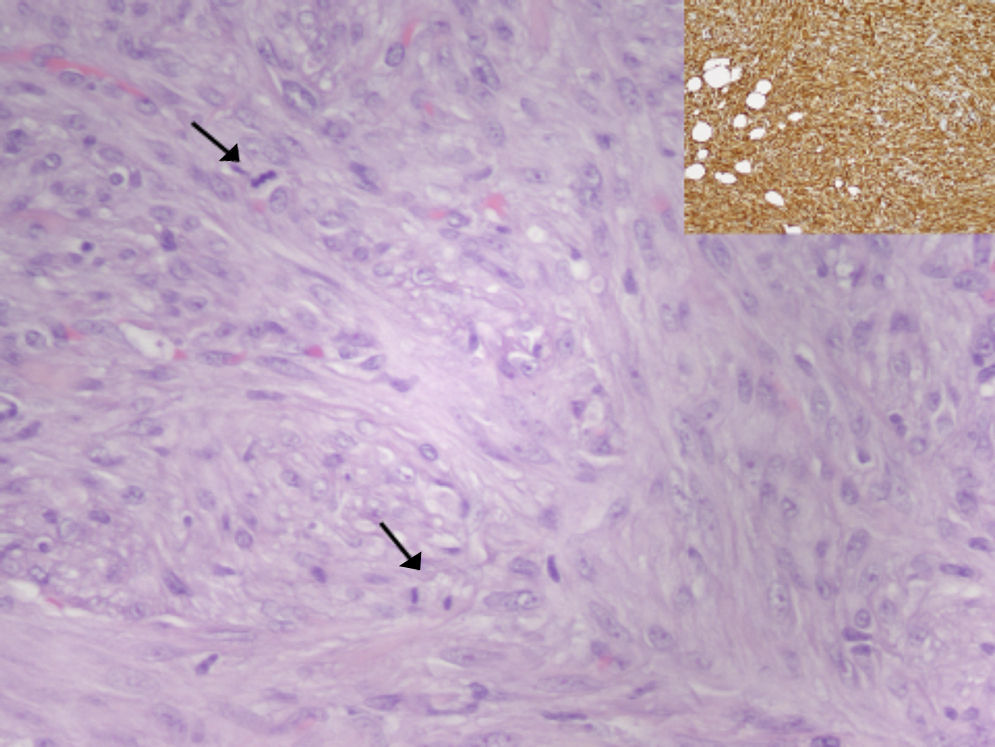
El análisis histológico de la pieza quirúrgica corresponde a una proliferación mesenquimal de patrón estoriforme y fascicular, constituida por células fusocelulares, con presencia de mitosis (15/10 campos de gran aumento) y de necrosis focal (aproximadamente 5%). Tras estudio inmunohistoquímico se evidencia tinción con vimentina y tinción focal con alfa 1-antitripsina y CD68. La tinción con marcadores epiteliales, musculares, neurales y vasculares resultó negativa. El índice de proliferación celular marcado con Ki67 fue aproximadamente del 35%. La tumoración medía 7cm de diámetro máximo y no alcanzaba los bordes de resección quirúrgicos. Tras valorar estos hallazgos, el diagnóstico fue de sarcoma pleomórfico indiferenciado (histiocitoma fibroso maligno pleomórfico), según la clasificación de tumores de partes blandas de la OMS de 2002 (fig. 2).
. Recuadro: la tinción con vimentina evidencia la naturaleza mesenquimal de la tumoración.")
Tras estudio del caso en sesión clínica multidisciplinar se decide administrar 3 ciclos de quimioterapia adyuvante con epirubicina-ifosfamida a dosis altas por tratarse de un tumor de alto grado.
Actualmente se encuentra en seguimiento por el Servicio de Oncología y Traumatología, así como por la Unidad del dolor para control del síndrome de dolor de miembro fantasma, pautándose pregabalina 150mg 1/12h y tramadol 50mg cada 8h, con buen control sintomático. El paciente es derivado al Servicio de Rehabilitación para valoración y se decide comenzar proceso de protetización por cumplir criterios adecuados para la realización de la misma, como son la edad joven del paciente, un muñón bien conformado sin alteraciones en la alineación y sin dolor a la palpación, un apoyo monopodal estable y la ausencia de asociación de otras patologías concomitantes, entre otros.
Los sarcomas de partes blandas (SPB) constituyen un grupo heterogéneo de neoplasias de origen mesenquimal, con excepción de los tumores de la vaina del nervio periférico, que son de origen ectodérmico. Constituyen el 88% del total de los sarcomas1. Representan el 1% de todos los tumores malignos del organismo y suelen presentarse como masas asintomáticas, motivo por el cual se retrasa su diagnóstico, presentando un 20% metástasis al ser identificadas, sobre todo a nivel pulmonar.
La edad media al diagnóstico es de 50-60años. El riesgo de aparición de estos tumores aumenta con la radioterapia previa por otras neoplasias, radiaciones ionizantes, linfedema crónico (linfangiosarcoma), exposición a cloruro de vinilo, arsénico, dioxinas y al contraste radiológico Thorotrasto (angiosarcoma hepático). También se asocia con algunos síndromes hereditarios (Werner, Gadner, esclerosis tuberosa, Gorlin, Von Recklinghausen)2.
Más del 50% de los sarcomas de tejidos blandos aparecen en cualquiera de los tejidos mesodérmicos de extremidades (solo un 14% en extremidades superiores), siendo el subtipo más frecuente el histiocitoma fibroso maligno o sarcoma pleomórfico (28%), seguido del liposarcoma (21%), el sarcoma sinovial (15%) y el leiomiosarcoma (12%). El resto los encontramos en el tronco y el retroperitoneo (40%) y en la región de cabeza y cuello (10%)1.
La forma de presentación más frecuente suele ser la de una tumoración de consistencia aumentada, indolora y de rápido crecimiento. Por eso, la atención primaria es uno de los ámbitos más propicios para la detección precoz de estas lesiones, teniendo siempre presentes los criterios que nos deben hacer pensar que se trata de una tumoración sospechosa. Estos criterios serían: tamaño superior a 5cm, consistencia aumentada, crecimiento rápido, localización profunda y masa recurrente tras extirpación previa3. Si estos criterios se cumplen, no debe retrasarse la derivación del paciente al ámbito hospitalario. El pronóstico principalmente depende del tamaño tumoral, del grado histológico y de la persistencia de tumor en los márgenes quirúrgicos. Los sarcomas de alto grado se relacionan con tasas más elevadas de fracaso local del tratamiento y potencial metastásico mayor4.
En caso se sospecha de SPB debe realizarse, en primer lugar, una resonancia magnética nuclear para valoración y extensión de la lesión, y toma de biopsia para planificar adecuadamente la cirugía5,6. El diagnóstico definitivo se realizará tras el análisis histológico de la pieza quirúrgica. Con el objetivo de conseguir una resección curativa, es importante planificar la biopsia inicial, para evitar complicaciones como las que presentó nuestro paciente. Si se confirma SPB de alto grado debe realizarse estudio de extensión con TAC para descartar metástasis.
El pronóstico depende principalmente del grado histológico (factor pronóstico más importante), subtipo histológico, tamaño tumoral, afectación de bordes quirúrgicos, localización (mejor pronóstico en extremidades), respuesta al tratamiento y si se trata de un tumor primario o de una recidiva (pronóstico más desfavorable)7,8.
El tratamiento va a depender de la histología del tumor, por lo que es fundamental que un patólogo experimentado revise el tejido biopsiado. El tratamiento se basa fundamentalmente en la cirugía (escisión completa de la lesión con márgenes adecuados)9 intentando preservar la funcionalidad, la radioterapia (tumores irresecables, alta probabilidad de tumor residual, márgenes menores de 2cm, amputación por resección extensa)1 y la quimioterapia, resultando más efectiva en tumores de alto grado, con doxorubicina e ifosfamida como fármacos más activos10.
En conclusión, los sarcomas son tumores poco frecuentes que debemos sospechar en nuestra consulta de atención primaria, tras una adecuada anamnesis y exploración física, ante la presencia de cualquier tumoración de partes blandas de características sospechosas de localización, principalmente, en extremidades inferiores. Un diagnóstico erróneo o un retraso en el mismo pueden suponer una importante disminución de la calidad de vida y de la supervivencia de estos pacientes.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
