






La enfermedad de Fabry (EF) es un raro trastorno lisosomal por déficit de la enzima alfa-galactosidasa A, con herencia ligada al cromosoma X, que ocasiona disfunción multiorgánica y muerte temprana. En 2004 se publicaron las características de los primeros 24 pacientes españoles incluidos en el registro Fabry Outcome Survey (FOS), habiendo aumentado significativamente su número desde entonces. Este trabajo analiza si ha habido cambios en el perfil clínico y diagnóstico de los mismos entre los 2 períodos.
Pacientes y métodoEn 2009 existían en el registro FOS datos de 92 pacientes. Se compararon los pacientes de 2003 con los posteriores (68) respecto a la edad de inicio de los síntomas y de diagnóstico, gravedad y diagnósticos previos erróneos, según sexo. Similar análisis se efectuó entre los casos índice (31) y el resto de pacientes.
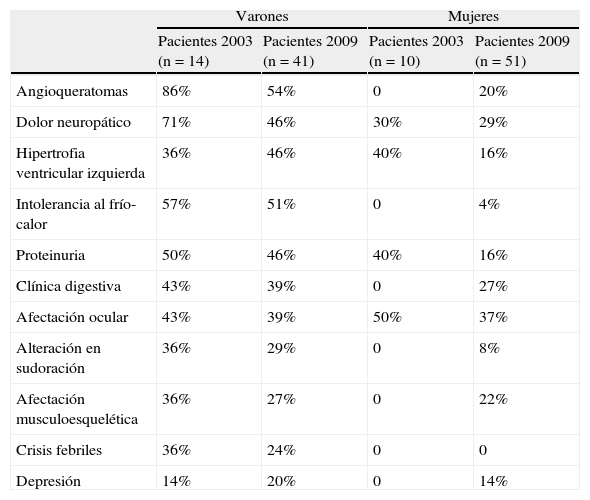
ResultadosEn ambos sexos la demora media del diagnóstico fue de 10 años. Los varones presentan un fenotipo clásico, y hasta un 40% de las mujeres referían síntomas. En estas, la actividad enzimática parece condicionar la gravedad de la enfermedad. No hubo diferencias en los parámetros analizados entre los pacientes del primer período y los posteriores, ni al comparar los casos índice con el resto.
ConclusionesLos registros como el FOS poseen valor para ahondar en el conocimiento de las enfermedades minoritarias. Confirmamos que las mujeres no son meras portadoras de la EF. Todavía falta formación y difusión para incluir la EF en el diagnóstico diferencial de otros procesos. Realizar estudios familiares exhaustivos ayudaría a un diagnóstico más precoz.
Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by a deficiency of the enzyme alpha-galactosidase A, that leads to multiorgan dysfunction and premature death. Data from the first 24 Spanish patients enrolled on the Fabry Outcome Survey (FOS) were published in 2004, with a significant increase in the number of patients since then. This manuscript analyzes whether the clinical profile or diagnosis of these patients between the 2 periods has changed.
Patients and methodsIn 2009 the FOS included data from 92 patients. Patients included up to 2003 and those included after that year (68) were compared by sex, regarding age at onset of symptoms and diagnosis, severity and previous misdiagnoses. Similar analysis was performed between the index cases (31) and the other patients.
ResultsMean delay in diagnosis was 10 years for both sexes. Male had a classic phenotype, and up to 40% of the females reported symptoms. In females, the enzyme activity seemed to determine disease severity. No differences were observed in any parameter when comparing the patients included in the first period to those included afterwards, nor when comparing index cases with the rest of the patients.
ConclusionsRegistries like FOS have a great value to deepen our understanding of rare diseases. We confirm that women are not just carriers of the disease. There is still a lack of education and awareness in order to include FD in the differential diagnosis of other processes. Complete family studies would allow early diagnosis of this disorder.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
