



La leucemia linfocítica crónica es un padecimiento maligno caracterizado por proliferación y acumulación de linfocitos B de aspecto maduro en médula ósea, sangre, ganglios linfáticos y en bazo. Como parte de su proceso de diferenciación, las moléculas de inmunoglobulina de las células B, además de los reordenamientos característicos en sus cadenas pesadas y ligeras, presentan un conjunto de mutaciones somáticas que incrementan enormemente su diversidad. La presencia o ausencia de mutaciones somáticas en las regiones variables del gen de las inmunoglobulinas en los linfocitos B malignos define la agresividad y el pronóstico del padecimiento en individuos con leucemia linfocítica crónica.
Chronic lymphocytic leukaemia is a malignant disease characterised by proliferation and accumulation of mature B lymphocytes in bone marrow, blood, lymph nodes, and spleen. As part of its process of differentiation, immunoglobulin molecules of B cells, in addition to the characteristic rearrangements in their heavy and light chains, present a set of somatic mutations that greatly increase their diversity. The presence or absence of somatic mutations in the variable regions of the immunoglobulin gene in the malignant B-cells defines the aggressiveness and prognosis of the disease in individuals with chronic lymphocytic leukaemia.
La leucemia linfocítica crónica (LLC) es un padecimiento causado por la acumulación progresiva de células B monoclonales, relativamente inmaduras e inmunológicamente incompetentes, en médula ósea, sangre, ganglios linfáticos y bazo1. Estas células B presentan un inmunofenotipo caracterizado por una alta expresión de CD20, CD22, CD43, CD19, CD23 CD79a, IgM/IgD y una ligera expresión de FMC7 y CD79b2-4. El desarrollo y la gravedad del padecimiento suelen ser muy variables, desde pacientes en los que la enfermedad progresa muy rápidamente hasta aquellos con enfermedad indolente que no requieren terapia5.
Actualmente se describe la LLC como un espectro de formas clínico-biológicas. Análisis de las cadenas de inmunoglobulinas (Ig) en linfocitos B indican que el 60-65% de las LLC muestran evidencia de hipermutación somática en las regiones variables de sus cadenas pesadas (IGHV), lo que puede modificar su afinidad a los antígenos; el 35-40% de las LLC carecen de mutaciones somáticas en las regiones IGHV6. El análisis del segmento génico que codifica para las regiones IGHV ha ayudado a comprender algunos aspectos de la enfermedad1. Los pacientes que no presentan mutaciones en IGHV muestran una enfermedad más agresiva con supervivencia más corta que aquellos pacientes que presentan tales mutaciones7.
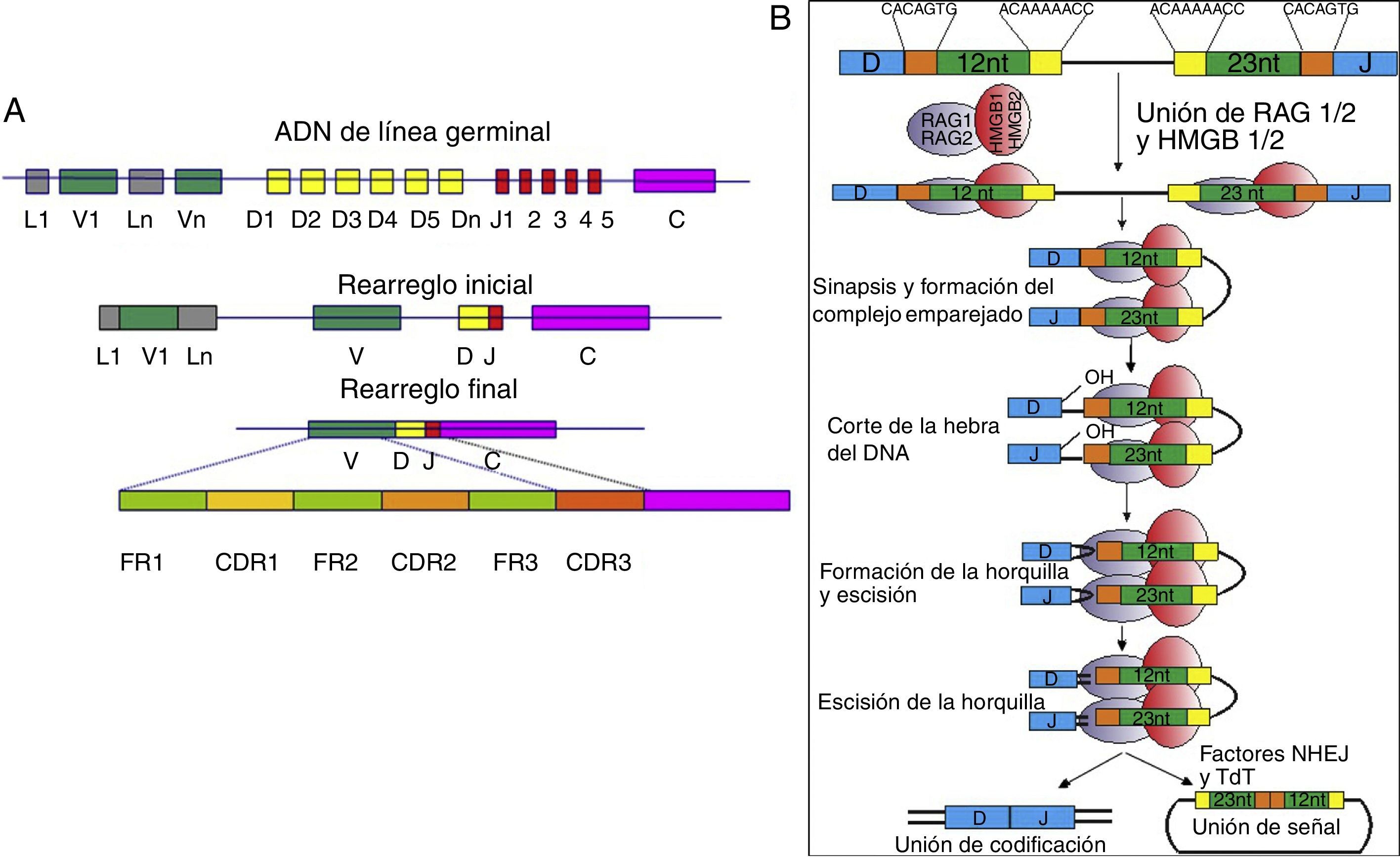
Reordenamientos en las secuencias génicas e hipermutación somática de las inmunoglobulinasLos exones de los genes de la región variable de las Ig se generan a través de un reordenamiento de secuencias que originalmente se encuentran separadas en la cadena de ADN original8. Las proteínas RAG1 y RAG2 son indispensables en el proceso de reordenamiento de los segmentos variables (V), de diversidad (D) y de unión (J) («recombinación VDJ») para formar la cadena pesada de la Ig (IgH)9.
El reordenamiento de las secuencias génicas de la IgH comienza en los linfocitos pro-B por acción de la enzima deoxinucleotil transferasa terminal (TdT) y los productos proteicos de los genes activadores de recombinación 1 y 2 (RAG1 y RAG2), con las que se inicia el reordenamiento inicial de los segmentos D y J de la IgH9. Posteriormente, en el estado de linfocitos pre-B I ocurre la unión de uno de los segmentos V al rearreglo inicial DJ, generándose el rearreglo final VDJ10,11 (fig. 1). Si el reordenamiento no da lugar a una IgH funcional, se iniciará el reordenamiento del segundo alelo de la IgH7.
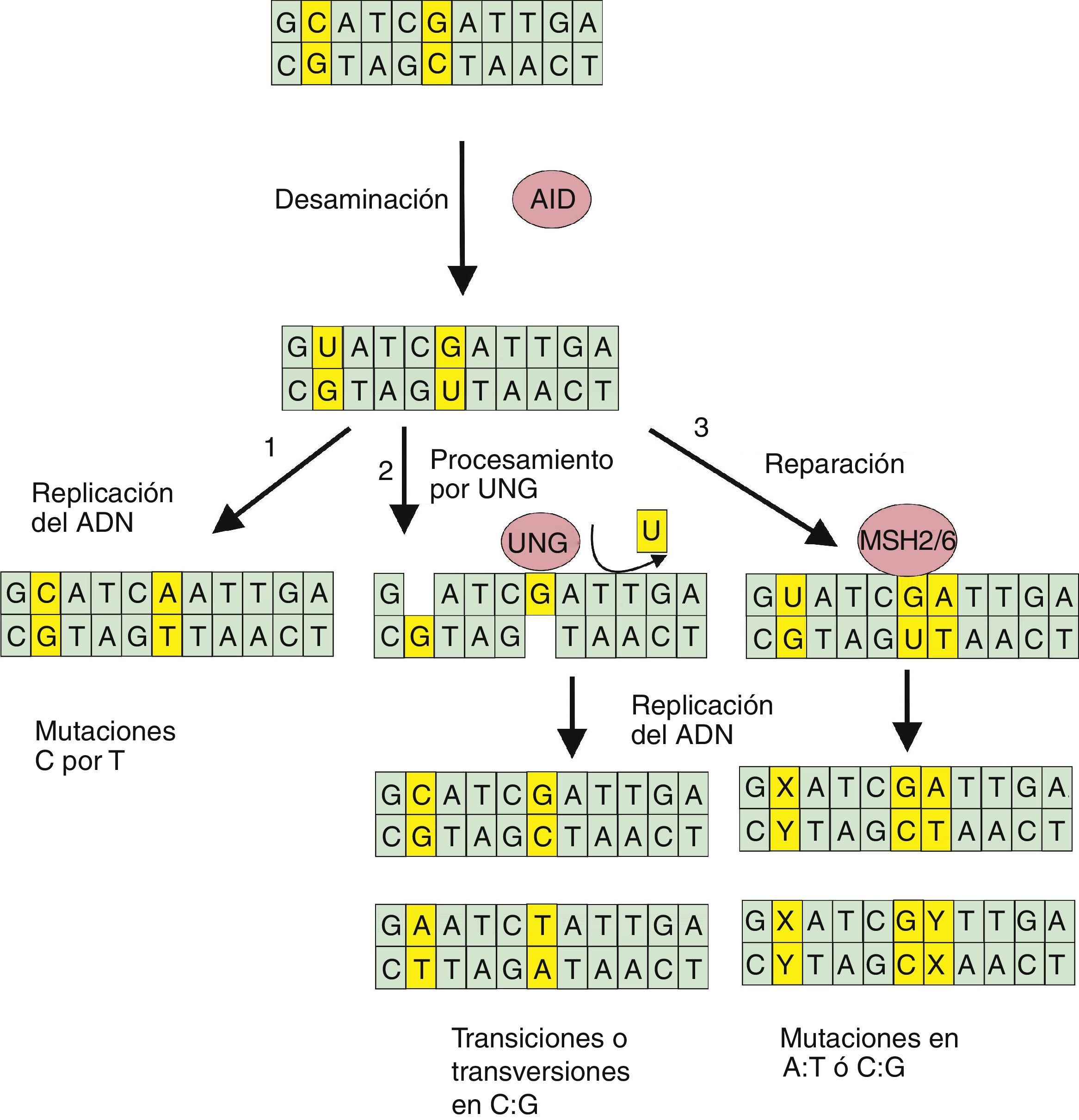
 la replicación del ADN conteniendo el uracilo no procesado genera mutaciones C→T; 2) la escisión de los uracilo por la enzima UNG genera sitios abásicos, mismos que tras la replicación del ADN permiten una gama más amplia de mutaciones de transición o transversión; 3) las proteínas MHS2/6 llevan a cabo un proceso de reparación mutagénico generando cambios en los pares de base A:T o C:G.")
Proceso de hipermutación somática. Luego de que la enzima AID convierte la citosina en uracilo en una de las hebras del ADN, el proceso puede continuar en varias formas: 1) la replicación del ADN conteniendo el uracilo no procesado genera mutaciones C→T; 2) la escisión de los uracilo por la enzima UNG genera sitios abásicos, mismos que tras la replicación del ADN permiten una gama más amplia de mutaciones de transición o transversión; 3) las proteínas MHS2/6 llevan a cabo un proceso de reparación mutagénico generando cambios en los pares de base A:T o C:G.
Posteriormente, y ya en los centros germinales, la célula B naïve (virgen) pasa por un proceso de hipermutación somática, que genera una enorme diversificación de Ig (fig. 2). Este proceso se inicia con la enzima denominada «desaminasa inducida por activación» (AID) que cataliza la desaminación selectiva de residuos de desoxicitidina en el ADN, transformándolos en residuos de uracilo. Los procesos destinados a la reparación de las modificaciones o lesiones U:G provocadas por la AID dan lugar a una variedad de sustituciones de bases en el ADN de las regiones IGHV, lo que constituye el llamado proceso de hipermutación somática. Así, la replicación del ADN provoca mutaciones de transición en los pares C:G, mientras que la enzima de escisión de uracilo (UNG) y la ADN glicosilasa crean sitios abásicos que pueden producir transversiones. Así mismo, el reconocimiento de los errores de apareamiento U:G por las proteínas MSH2/MSH6 desencadena un proceso de reparación mutagénico en el que la polimerasa η desempeña un papel importante y conduce a mutaciones en los pares A:T. En resumen, la desaminación del ADN provocada por la enzima AID permite la conversión de las secuencias variables de las Ig, el cambio de clase de isotipo y algunas translocaciones oncogénicas en tumores de células B12 (fig. 2).
 Reordenamiento de los segmentos génicos de la cadena original del gen de las cadenas pesadas de la inmunoglobulina. El rearreglo inicial logra la reunión de los segmentos D-J y el rearreglo final la de los segmentos V-D-J. El rearreglo final está conformado por secuencias constantes denominadas marcos (FR) y secuencias variables denominadas regiones determinantes de complementariedad (CDR). B) Mecanismo molecular de recombinación de las regiones V-D-J. Las proteínas RAG1/2 (óvalos morados) junto con las proteínas HMGB1/2 (óvalos rojos) se unen a la secuencia de heptámeros y nonámeros; las proteínas HMGB1/2 le confieren flexibilidad al ADN y las proteínas RAG1/2 cortan una sola hebra del ADN generando un extremo 3’OH libre, el cual se une covalentemente al enlace fosfodiéster de la cadena opuesta, formando una horquilla en el extremo de codificación y una señal 5’ fosforilada; los extremos permanecen asociados al complejo proteico denominado complejo de postescisión. Finalmente, los extremos en horquilla se procesan con ayuda del complejo formado por la proteína de «unión final no homologa» (NHEJ) y la deoxinucleotil transferasa terminal (TdT) para crear el rearreglo inicial de la inmunoglobulina43,44.")
A) Reordenamiento de los segmentos génicos de la cadena original del gen de las cadenas pesadas de la inmunoglobulina. El rearreglo inicial logra la reunión de los segmentos D-J y el rearreglo final la de los segmentos V-D-J. El rearreglo final está conformado por secuencias constantes denominadas marcos (FR) y secuencias variables denominadas regiones determinantes de complementariedad (CDR). B) Mecanismo molecular de recombinación de las regiones V-D-J. Las proteínas RAG1/2 (óvalos morados) junto con las proteínas HMGB1/2 (óvalos rojos) se unen a la secuencia de heptámeros y nonámeros; las proteínas HMGB1/2 le confieren flexibilidad al ADN y las proteínas RAG1/2 cortan una sola hebra del ADN generando un extremo 3’OH libre, el cual se une covalentemente al enlace fosfodiéster de la cadena opuesta, formando una horquilla en el extremo de codificación y una señal 5’ fosforilada; los extremos permanecen asociados al complejo proteico denominado complejo de postescisión. Finalmente, los extremos en horquilla se procesan con ayuda del complejo formado por la proteína de «unión final no homologa» (NHEJ) y la deoxinucleotil transferasa terminal (TdT) para crear el rearreglo inicial de la inmunoglobulina43,44.
Como neoplasia maligna de células B, la LLC puede evolucionar a partir de células B en estado pregerminal o posgerminal. Esto divide las LLC en 2 subconjuntos: 1) leucemias cuyas células B han pasado con éxito a través del centro germinal y han completado los rearreglos de la cadena original y los procesos de hipermutación somática de la Ig13-15. Los pacientes que presentan linfocitos B con regiones IGHV mutadas tienen una mayor frecuencia de deleciones 13q14 (65% vs. 48% p=0.004)16, y 2) leucemias que se originan a partir de linfocitos B que no han completado los reordenamientos de línea germinal o el proceso de hipermutación somática, en las que el evento mutacional se habría iniciado en linfocitos tipo pro-B o en células B naïve13-15. Este tipo de células tienen, con mayor frecuencia, aberraciones genómicas de mal pronóstico como la deleción 17p13 (10% vs. 3%, p=0.03) o la deleción 11q23 (27% vs. 4%, p<0.001)16. Para analizar la variación de las regiones IGHV se considera que las secuencias que tienen menos de 2% de homología con respecto al ADN de línea germinal ya han pasado por el proceso de hipermutación somática17. Se conoce que la actividad transcripcional de las regiones IGHV está regulada por proteínas específicas, como: PTEN, FOS, TNF, IFGR2, etc.18. La supervivencia media de pacientes con LLC, cuyos linfocitos no muestran hipermutación somática, es de 95 meses en comparación con 293 meses en pacientes con LLC cuyos linfocitos sufrieron el proceso mutacional (p<0.001)7,19. Esta diferencia hace que el análisis de las secuencias IGHV sea importante con propósitos de pronóstico de los pacientes con LLC.
Los segmentos génicos IGHV presentes en las células de LLC difieren de los linfocitos B CD5+ normales. Así, los linfocitos de LLC utilizan predominantemente segmentos génicos de las familias HV1, HV3 y HV4, en una distribución que es diferente de la reportada para los linfocitos B normales CD5+20,21. Específicamente, en linfocitos de pacientes con LLC la familia HV1 se sobreexpresa, en tanto que la familia HV3 muestra baja expresión en comparación con los linfocitos normales. Las secuencias génicas utilizadas con más frecuencia dentro de estas familias son: HV1-69, HV3-07, HV3-23 y HV4-34, aunque la frecuencia relativa en la que estos segmentos génicos se muestran en diferentes cohortes de LLC varía20,22,23.
Es importante destacar que las mutaciones somáticas no parecen producirse con frecuencia uniforme entre los subgrupos de segmentos génicos IGHV, sino que muestran una jerarquía de frecuencias (IGHV3>IGHV4>IGHV1)20. Por ejemplo, los segmentos génicos HV3-07, HV1-69 y HV4-34 se encuentran más frecuentemente en LLC-B que en la población general de células B normales o CD5+. Los segmentos génicos HV3-07, HV3-23 se encuentran más frecuentemente en células mutadas, mientras que HV1-69 en la mayoría de los casos se encuentra en células no mutadas24. Para HV4-34 la distribución es similar en las 2 formas25.
Linfocitos de leucemia linfocítica crónica sin mutaciones en las regiones variables de las cadenas pesadas de la inmunoglobulinaEl proceso de hipermutación somática en los genes de las Ig implica cierto grado de estimulación/selección de los linfocitos en maduración, por lo que es necesario entender la existencia de LLC con regiones IGHV no mutadas. Un modelo explica el origen de estas células no mutadas como derivadas de linfocitos que residen en la zona marginal y allí reciben estimulación antigénica. Por lo tanto, es probable que las células madre que originan este tipo de leucemias B provengan de la proliferación de células vírgenes o que hayan sufrido estimulación fuera del centro germinal por antígenos T-independientes (como es el caso de los polisacáridos o lipopolisacáridos), los cuales inducen dicha proliferación sin acumulación de mutaciones en las regiones IGHV21 (fig. 3).
 LLC con regiones IGHV no mutadas: 1) Estimulación mediante antígeno (Ag) independiente de células T; 2) activación del linfocito B, y 3) evento neoplásico. B) LLC con regiones IGHV mutadas: 1) activación de la célula B naïve dependiente de linfocito T; 2) expansión clonal e hipermutación somática; 3) estimulación del linfocito B por el linfocito T cooperador (TH); 4) el linfocito B se activa y pasa a linfocito B de memoria, 5) evento neoplásico.")
A) LLC con regiones IGHV no mutadas: 1) Estimulación mediante antígeno (Ag) independiente de células T; 2) activación del linfocito B, y 3) evento neoplásico. B) LLC con regiones IGHV mutadas: 1) activación de la célula B naïve dependiente de linfocito T; 2) expansión clonal e hipermutación somática; 3) estimulación del linfocito B por el linfocito T cooperador (TH); 4) el linfocito B se activa y pasa a linfocito B de memoria, 5) evento neoplásico.
Los receptores de células B (BCR) poliespecíficos serían la primera línea de defensa contra patógenos. Se ha visto que los BCR en algunas LLC presenta reminiscencias estructurales de anticuerpos que han reaccionado contra polisacáridos capsulares de antígenos bacterianos21,26. Esto, unido a las observaciones logradas en ratones, en los que las células de la zona marginal median la respuesta de antígenos T independientes, sugiere que las células progenitoras podrían derivar de este conjunto. En principio, la formación del repertorio BCR y la transformación maligna pueden ocurrir conjunta o secuencialmente. La transformación también puede comenzar cuando la célula está siendo estimulada, o una célula con un BCR determinado puede transformarse con posterioridad. Repetidas observaciones muestran que en algunas células de LLC la estimulación antigénica continúa durante la transformación27,28.
En general, la hipótesis del origen de las células no mutadas en el precentro germinal (CG) es factible y concordante con los datos disponibles actualmente, pero no logra explicar por qué los perfiles de expresión de células de LLC mutadas muestran solo pequeñas diferencias entre sí, a pesar de sus presuntos programas funcionales y orígenes celulares diferentes29,30.
Linfocitos de leucemia linfocítica crónica con mutaciones en las regiones variables de las cadenas pesadas de la inmunoglobulinaSe ha propuesto un modelo por el cual los linfocitos con regiones IGHV mutadas se originan de linfocitos B de memoria que han abandonado el CG, dado que la presencia de mutaciones es un marcador de células B post-CG30. En general, las células de LLC no muestran mutaciones progresivas, como en el caso del linfoma folicular, en el que todas las transformaciones ocurren en el CG y sus células muestran un proceso muy activo de diversificación de los genes IGHV. Además, las células de LLC mutadas también presentan mutaciones en el gen BCL-6, como la mayoría de células B post-CG. Este modelo plantea ciertas diferencias, por ejemplo que en la mayoría de las células B post-CG el isotipo predominante es IgG, mientras que las células de LLC expresan en su mayoría IgM e IgD31,32 (fig. 3).
En otro escenario se plantea la posibilidad de que, tanto las células mutadas como no mutadas, derivan de una célula B de la zona marginal IgM+, IgDlow (fig. 3). Estas células responden a antígenos T-independientes y pueden tener IGHV mutado o no mutado30. Existen evidencias, aunque indirectas, de que estas células pueden ser estimuladas por bacterias, antígenos virales o antígenos propios dentro de la zona marginal y acumular mutaciones fuera del CG21. Además, las células de LLC mutadas y no mutadas derivarían de la misma célula, aunque quizás en diferentes estadios de maduración, lo cual estaría en concordancia con los estudios de perfiles de expresión similar entre ambos grupos29,30.
En una tercera línea de posibilidades, y aceptando que la estimulación pueda darse por un antígeno T independiente, los linfocitos de la zona marginal podrían ir a los órganos linfoides y ayudar a crear los CG donde ocurre el proceso de mutación, como sucede en ratones. En este modelo la transformación podría ocurrir después de la salida de las células del CG21.
En estos últimos modelos, si los progenitores son células B de la zona marginal IgM+ e IgDlow, el proceso de activación al que son expuestas las células de LLC, tanto por estimulación por antígenos propios y/o transformación leucémica, podrían inducir a la aparición de nuevos marcadores de superficie. Así, la activación de células quiescentes de la zona marginal induce la expresión de moléculas de activación como CD5, CD23 y CD3821.
Correlación entre CD38, ZAP-70 y el estado mutacional de las regiones variables de las cadenas pesadas de la inmunoglobulinaLa definición de LLC mutada y no mutada se da por un límite establecido arbitrariamente de 98% de homología con el gen de la línea germinal. En general, el fenotipo leucémico de peor pronóstico se asocia típicamente con la presencia de regiones IGHV no mutadas7, con expresión incrementada del marcador de superficie CD3819 y de la proteína ZAP-7033, y con la presencia de aberraciones cromosómicas como las deleciones 17p13 (proteína p53) y 11q23 (ataxia telangiectasia ATM)34, mientras que el fenotipo de mejor pronóstico se asocia a linfocitos que presentan regiones IGHV mutadas, baja expresión de CD38 y ZAP-70, y cariotipo normal o con deleciones 13q1435,36.
Se ha visto que los pacientes de LLC con regiones IGHV no mutadas presentan una mayor expresión de CD38 (≥ 30%) en comparación con aquellos que presentan regiones IGHV mutadas; por el contrario, la disminución en la expresión de CD38 (< 30%) se asocia a un genotipo de regiones IGHV mutadas36. Damle et al. demostraron que los resultados del tratamiento para pacientes con regiones IGHV no mutadas y pacientes mostrando expresión aumentada de CD38 (≥ 30%) fueron similares; es decir, el 79.2% de los pacientes con IGHV no mutado y el 76.5% de los pacientes con ≥ 30% CD38 requirieron tratamientos continuos. Por el contrario, el 52.2% con los pacientes que presentan linfocitos con regiones IGHV mutadas y el 73% de los pacientes con expresión disminuida de CD38 (< 30%) no requirieron terapia o fue mínima19.
El impacto negativo de la sobrexpresión de CD38 en la evolución y supervivencia de estos pacientes también fue demostrado por del Poeta et al., quienes encontraron que una expresión de CD38 ≥ 30% en el momento del diagnóstico se asoció con etapas de rai intermedias/altas37. La expresión de CD38 y el estado mutacional de IGHV son factores de riesgo independientes que podrían ser utilizados para la estadificación clínica37,38.
Del mismo modo, Rassenti et al. encontraron que la expresión de la proteína ZAP-70+ (≥ 20%) en el 71% de los casos de LLC se encontraba asociada con regiones IGHV no mutadas, pero solo con el 17% de los casos de IGHV mutadas (p<0.001). Estos autores sugieren que ZAP-70 puede ser un predictor más importante que la presencia de regiones IGHV no mutadas, para determinar el inicio de la terapia en pacientes con LLC39.
Mediante cuantificación en tiempo real (RQ-PCR) de la expresión de ZAP-70 se demostró que el 94% de los pacientes con LLC que presentaban regiones IGHV no mutadas fueron ZAP-70+ y el 92% de los pacientes con IGHV mutadas fueron ZAP-70–40. Sin embargo, algunos estudios sugieren que la expresión de ZAP-70 puede cambiar con el tiempo, particularmente en el momento de la progresión de la enfermedad41,42.
ConclusionesMarcadores biológicos como el estado mutacional de las regiones IGHV y la expresión de CD38 y ZAP-70 parecen ser eficientes indicadores pronósticos para ayudar a definir una mejor estadificación de los casos de LLC y lograr que los pacientes reciban el mejor y más oportuno tratamiento para su enfermedad. Los pacientes con LLC, cuyos linfocitos muestran regiones IGHV no mutadas, presentan un padecimiento más agresivo, puesto que el evento neoplásico o transformante tiene lugar en etapas más tempranas de su evolución, probablemente en precentros germinales.
Seguramente nuevos y mayores estudios permitirán saber en qué momento el linfocito B pasa a ser una célula neoplásica, entender el papel que desempeña el antígeno en este proceso de evolución y comprender a cabalidad las vías que sigue el linfocito B en el proceso de hipermutación somática.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés con respecto a este manuscrito.