




Las herramientas genotípicas basadas en el análisis de la región V3 de la envuelta viral se perfilan como la alternativa a los ensayos fenotípicos para la determinación del tropismo del VIH por los receptores de quimiocinas CCR5 y CXCR4 en la práctica clínica. Este trabajo evalúa la concordancia entre los distintos algoritmos de interpretación genotípica actualmente disponibles en pacientes VIH infectados con subtipo B versus subtipos no-B.
MétodosSe seleccionaron pacientes VIH positivos, procedentes del Complexo Hospitalario Universitario de Santiago de Compostela (CHUS), España. A partir de muestras de plasma, se amplificó y secuenció la región V3 de la envuelta viral. El tropismo viral se determinó usando 8 algoritmos genotípicos distintos. La concordancia entre los distintos predictores se evaluó calculando el índice de concordancia kappa. El subtipo genético fue determinado por análisis filogenético.
ResultadosSe incluyeron un total de 92 pacientes, 72 infectados por subtipo B y 20 por no-B. En pacientes con subtipo B, se obtuvieron valores significativos de kappa para todas combinaciones posibles (n=28) entre los algoritmos genotípicos analizados. La mejores valores entre predictores no relacionados se obtuvieron para webPSSMSINSI/WetcatPART (k: 0,771) y webPSSMSINSI/geno2pheno (k: 0,574). En subtipos no-B solo se obtuvieron valores significativos para 13 combinaciones, correspondiendo los mejores a PSSMX4R5/WetcatPART (k: 0,600) y PSSMSINSI/Charge rule (k: 0,590).
ConclusiónExiste una alta concordancia entre los distintos algoritmos genotípicos utilizados para la determinación del tropismo viral en pacientes infectados con subtipo B, especialmente con webPSSMSINSI y geno2pheno o Wetcat. Sin embargo, la concordancia general de los algoritmos para subtipos no-B es menor. Esta falta de consenso podría justificarse por la baja prevalencia de los subtipos no B tienen en las bases de datos empleadas para el entrenamiento de los predictores genotípicos.
Genotypic tools based on the analysis of the V3 region are seen as an alternative to phenotypic assays for viral tropism determination before prescribing maraviroc. The concordance between different genotypic algorithms has been evaluated in HIV+ patients infected with B versus non-B subtypes.
MethodsHIV-infected patients on regular follow up at Hospital Universitario de Santiago de Compostela (Spain) were selected. The env-V3 region was sequenced from plasma samples and viral tropism was estimated using 8 different genotypic algorithms. Concordance among predictors was statistically evaluated by the calculation of the kappa index. Phylogenetic analyses were performed to determine the genetic subtype.
ResultsA total of 92 HIV-infected patients were selected, 72 B and 20 non-B subtypes. Regarding the B subtype group, significant kappa values were obtained among all 28 possible combinations between the genotypic predictors evaluated. The best concordance among non-related predictors was observed for webPSSMSINSI/WetcatPART (k: 0.771) and webPSSMSINSI/geno2pheno (k: 0.574). Conversely, among non-B subtypes, a significative kappa index was only obtained for 13 combinations. Among non-B subtypes, the best concordance values were obtained for webPSSMX4R5/WetcatPART (k: 0.600) and webPSSMSINSI/Charge rule (k: 0.590).
ConclusionA high concordance was observed between different genotypic algorithms to determine viral tropism among HIV-1 B subtypes infected patients, especially between webPSSMSINSI and geno2pheno or Wetcat. Conversely, the overall concordance among non-B subtypes was lower. This heterogeneity could be justified by the low prevalence of non B subtypes in the datasets in which the genotypic tropism predictors were trained.
En los últimos años, los estudios de tropismo por los receptores de quimiocinas CCR5 y CXCR4 utilizados mayoritariamente por el VIH-1 para entrar en la célula han adquirido un especial interés debido al desarrollo clínico y aprobación de nuevos fármacos inhibidores de la entrada diseñados específicamente para bloquear la unión de la glicoproteína gp120 de la envuelta viral con estos correceptores1-3. La familia de los antagonistas de los correceptores está representada hasta el momento por el antagonista de CCR5, maraviroc (Celsentri®)4. Los antagonistas de CCR5 inhiben de forma selectiva la replicación de variantes virales R5 trópicas5,6. Por tanto, antes de la prescripción de estos fármacos es necesaria la determinación del tropismo viral con el fin de asegurar una óptima respuesta clínica al fármaco7,8.
Los ensayos fenotípicos han sido considerados los métodos más fiables para la determinación del tropismo viral9. Más concretamente, el ensayo fenotípico de Trofile® (Monogram Biosciences, San Francisco, CA, EE.UU.)10, utilizado ampliamente en los ensayos clínicos con antagonistas de CCR5 y posteriormente su versión mejorada ESTA-Trofile® (Enhanced Sensitivity Trofile Assay)11 ha sido en general el más empleado para la determinación del tropismo en la práctica clínica. Sin embargo, el ensayo de Trofile®, presenta limitaciones técnicas y logísticas que lo sitúan lejos de la herramienta diagnóstica ideal para su utilización en la práctica clínica, lo que supone en muchas ocasiones una barrera entre el clínico y la prescripción del fármaco.
Los ensayos genotípicos basados en el análisis de la región V3 de la envuelta viral se presentan como una alternativa más económica, rápida, y factible de desarrollar en laboratorios especializados en el diagnóstico de la infección por VIH. Existen diferentes reglas y algoritmos de interpretación del tropismo viral basados fundamentalmente en las características de la secuencia de aminoácidos de la región V3 de la envuelta viral12-14.
En los últimos años, se ha evaluado la fiabilidad de las herramientas genotípicas para la determinación del tropismo viral en muestras clínicas. Para ello, se han desarrollado diferentes estudios para establecer la concordancia entre herramientas genotípicas y fenotípicas. En un principio, los resultados obtenidos generalmente coincidían en que los ensayos genotípicos presentaban una alta especificidad, pero baja sensibilidad para la identificación de variantes X4-trópicas15,16. Posteriormente, y gracias a la implementación de mejoras en la interpretación de los algoritmos genotípicos se ha conseguido incrementar considerablemente su sensibilidad17-19. Sin embargo, la validación de las herramientas genotípicas para uso clínico requiere la demostración de su capacidad para identificar pacientes respondedores y no respondedores a un tratamiento con antagonistas de CCR5, más que demostrar una buena correlación con el ensayo de Trofile®. Recientemente, Harrigan et al20,21, en análisis retrospectivos de los ensayos MOTIVATE y MERIT, han demostrado resultados comparables en la predicción de respuesta a maraviroc entre algunas herramientas genotípicas (geno2pheno y webPSSM) y el ensayo de Trofile®. Además, en una cohorte de pacientes de Berlín se ha demostrado de forma prospectiva la capacidad de la herramienta genotípica geno2pheno de predecir respuesta a maraviroc de forma similar a Trofile®22. Esta capacidad no está demostrada de forma prospectiva para el resto de algoritmos genotípicos, por lo que resultaría de gran interés, conocer la concordancia y correlación entre las diferentes herramientas genotípicas, especialmente entre geno2pheno y los demás algoritmos de interpretación genotípica. Además, la introducción de subtipos no-B del VIH-1 está experimentando en los últimos años un incremento en los países occidentales. Concretamente en España, se estima que la prevalencia de subtipos no-B del VIH-1 en nuevos diagnósticos es del 15-18%23,24. Por tanto, es necesario conocer la influencia del subtipo en las distintas herramientas genotípicas para la interpretación del tropismo viral.
Los objetivos del presente trabajo son determinar la concordancia existente entre la interpretación dada por los diferentes algoritmos de predicción genotípica del tropismo viral del VIH-1, y evaluar si dicha concordancia puede verse afectada por la influencia del subtipo viral.
Pacientes y métodosPoblación de estudioPara la realización de este estudio se utilizaron las muestras clínicas de plasma de pacientes que fueron diagnosticados de infección por VIH, entre septiembre de 2006 y mayo de 2009, en el Servicio de Microbiología del Complexo Hospitalario Universitario de Santiago (Santiago de Compostela, España), así como de pacientes en seguimiento de la infección por VIH-1 procedentes del mismo centro. Las muestras de plasma fueron almacenadas a -20°C hasta la realización de los correspondientes estudios genotípicos.
Determinación del subtipo viral del VIH-1Las muestras del estudio fueron caracterizadas genotípicamente en subtipos en base a la secuenciación directa y parcial (fragmento de 1.200 nt) del gen pol, que incluye la proteasa (PR) y los primeros 300 aminoácidos de la retrotranscriptasa viral (RT), y al posterior análisis filogenético de las secuencias obtenidas.
El análisis filogenético se realizó utilizando el paquete informático PHYLIP. El alineamiento con las secuencias de referencia correspondientes a los 9 subtipos principales y a las 43 formas recombinantes circulantes (CRFs) obtenidas de la base de datos de Los Alamos (http://www.hiv.lanl.gov), se realizó con el programa CLUSTALW. Las distancias genéticas fueron estimadas utilizando el programa DNAdist (método Kimura dos-parámetros) y las asociaciones filogenéticas se determinaron usando el método neighbor-joining (Neighbor).
Determinación de la predicción genotípica del correceptor del VIH-1La predicción genótipica del tropismo viral se realizó mediante el análisis genético de la región variable V3 de la glicoproteína de la envuelta gp120. Las secuencias de V3 de los virus que infectaban a cada paciente fueron amplificadas y posteriormente caracterizadas genotípicamente mediante secuenciación directa según condiciones ya descritas14. Las secuencias obtenidas fueron interpretadas como R5-trópicas o X4-trópicas conforme a 8 algoritmos genotípicos diferentes predictores del tropismo viral del VIH-1 en tres diferentes portales de internet:
- 1.
http://genomiac2.ucsd.edu:8080/wetcat/v3.html:C4.5, C4.5p8p12, PART, SVM, y Charge Rule.
- 2.
http://ubik.microbiol.washington.edu/computing/pssm/: webPSSMX4/R5 y webPSSMSI/NSI.
- 3.
http://coreceptor.bioinf.mpi-sb.mpg.de/cgi-bin/coreceptor.pl: geno2pheno[coreceptor] (FPR=5%).
Brevemente, todos los algoritmos genotípicos están fundamentados en bases de datos que contienen datos pareados de tropismo fenotípico y secuencias V3. Mediante diferentes métodos estadísticos se identifican posiciones críticas de la secuencia de V3 asociadas con el tropismo viral. Así, las matrices C4.5, C4.5p8p12 y PART son algoritmos basados en árboles de decisiones; WetcatSVM y geno2pheno son algoritmos basados en métodos estadísticos del tipo support vector machine-SVM; mientras que webPSSM utiliza un método estadístico del tipo position specific scoring matrices-PSSM. En el caso de la regla “charge rule”, se trata de un algoritmo más sencillo únicamente basado en el ánalisis de las posiciones 11 y 25 de la región V3.
Análisis estadístico de datosLa concordancia entre los diferentes algoritmos predictores del tropismo viral se realizó mediante tablas de contingencia determinando el índice de concordancia kappa (k) para variables cualitativas con un intervalo de confianza del 95% (IC 95%). El coeficiente kappa es un índice que incorpora el papel del azar en el cálculo de la concordancia. La valoración del índice kappa se hizo en base a la propuesta de Landis y Koch25, según la cual se consideraron 5 grados de concordancia: pobre (k entre 0 y 0,2), bajo (0,2-0,4), moderado (0,4-0,6), bueno (0,6-0,8) y muy bueno (0,8-1). No se consideraron los valores de kappa no significativos. Todas las pruebas de significación son bilaterales y se han considerado significativos solo aquellos valores de p<0,05. Tanto para la determinación del coeficiente kappa como para las demas determinaciones (chi-cuadrado de Pearson o test exacto de Fisher) realizadas con otras variables se utilizó el programa estadístico SPSS v15.0 (SPSS Inc, Chicago, IL,EE.UU.).
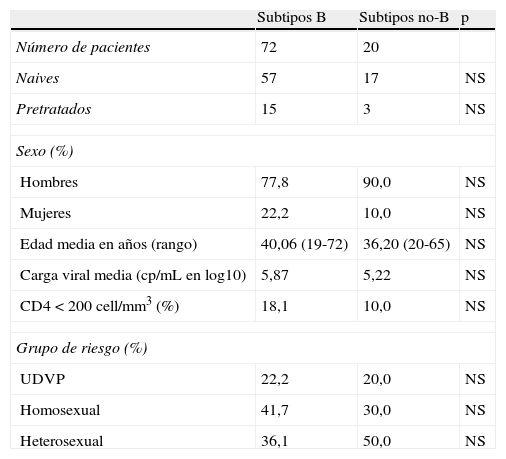
ResultadosCaracterísticas de la población de estudioSe obtuvieron muestras de plasma de 92 pacientes, 18 mujeres y 74 hombres. El 80,4% (74/92) habian sido diagnosticados de infección por VIH-1 durante el período de estudio, mientras que el 19,6% (18/92) corresponde a pacientes pretratados. Por lo que respecta al análisis filogenético para la determinación del subtipo genético viral, 72 pacientes pertenecían al subtipo B, mientras que 20 pacientes pertenecían a diferentes subtipos no-B distribuídos de las siguiente forma: 2C, 5 D, 5 F, 3 CRF01_AE, 1 CRF02_AG, 2 CRF12_BF y 2 CRF14_BG.
Las características de los pacientes se recogen en la tabla 1. Entre los grupos de pacientes infectados con subtipos B y no-B del VIH-1 no se encontraron diferencias significativas entre las distintas variables correspondientes a las características generales de la población de estudio.
Características generales de la población de estudio según el subtipo genético.
| Subtipos B | Subtipos no-B | p | |
| Número de pacientes | 72 | 20 | |
| Naives | 57 | 17 | NS |
| Pretratados | 15 | 3 | NS |
| Sexo (%) | |||
| Hombres | 77,8 | 90,0 | NS |
| Mujeres | 22,2 | 10,0 | NS |
| Edad media en años (rango) | 40,06 (19-72) | 36,20 (20-65) | NS |
| Carga viral media (cp/mL en log10) | 5,87 | 5,22 | NS |
| CD4<200 cell/mm3 (%) | 18,1 | 10,0 | NS |
| Grupo de riesgo (%) | |||
| UDVP | 22,2 | 20,0 | NS |
| Homosexual | 41,7 | 30,0 | NS |
| Heterosexual | 36,1 | 50,0 | NS |
El análisis de concordancia entre los diferentes algoritmos de predicción del tropismo viral se realizó en 2 grupos de pacientes por separado: aquellos infectados por variantes VIH-1 subtipo B versus pacientes infectados por VIH-1 subtipos no-B.
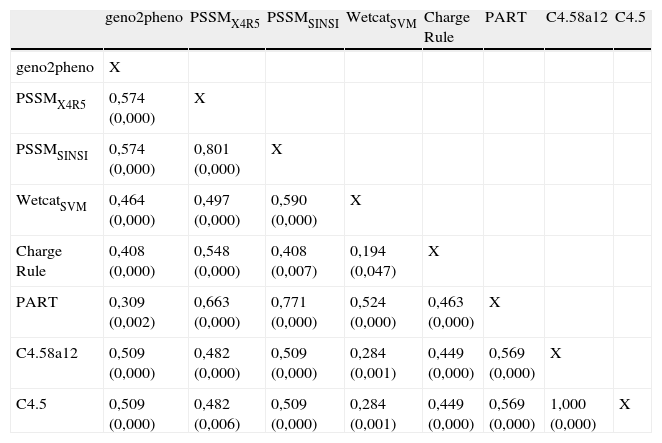
Tal como se observa en la tabla 2, dentro de la población de estudio de pacientes infectados por el subtipo B del VIH-1 encontramos una concordancia estadísticamente significativa entre todos los algoritmos estudiados. De las 28 posibles comparaciones, el índice kappa se clasificó como pobre en 1 caso, bajo en 3 casos, moderado en 20 casos, bueno en 2 casos y muy bueno en otros 2 casos. La mejor concordancia entre algoritmos no relacionados se observó para webPSSMSINSI/WetcatPART (k: 0,771), y webPSSMSINSI/geno2pheno (k: 0,574).
Concordancia entre los diferentes algoritmos de predicción genotípica del tropismo viral en pacientes infectados con VIH-1 subtipo viral B (índice de kappa y grado de significación).
| geno2pheno | PSSMX4R5 | PSSMSINSI | WetcatSVM | Charge Rule | PART | C4.58a12 | C4.5 | |
| geno2pheno | X | |||||||
| PSSMX4R5 | 0,574 (0,000) | X | ||||||
| PSSMSINSI | 0,574 (0,000) | 0,801 (0,000) | X | |||||
| WetcatSVM | 0,464 (0,000) | 0,497 (0,000) | 0,590 (0,000) | X | ||||
| Charge Rule | 0,408 (0,000) | 0,548 (0,000) | 0,408 (0,007) | 0,194 (0,047) | X | |||
| PART | 0,309 (0,002) | 0,663 (0,000) | 0,771 (0,000) | 0,524 (0,000) | 0,463 (0,000) | X | ||
| C4.58a12 | 0,509 (0,000) | 0,482 (0,000) | 0,509 (0,000) | 0,284 (0,001) | 0,449 (0,000) | 0,569 (0,000) | X | |
| C4.5 | 0,509 (0,000) | 0,482 (0,006) | 0,509 (0,000) | 0,284 (0,001) | 0,449 (0,000) | 0,569 (0,000) | 1,000 (0,000) | X |
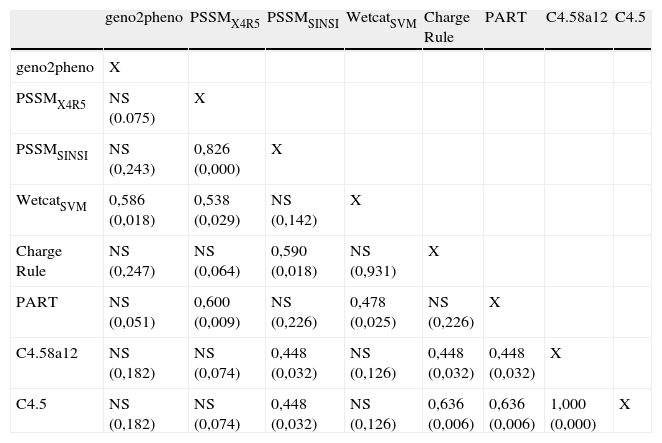
Sin embargo, dentro del grupo correspondiente a los subtipos no-B y tal como se observa en la tabla 3, de las 28 posibilidades de combinación entre los distintos algoritmos utilizados, en 15 no existía nivel de significación asociado a la concordancia. De los 13 restantes, el grado de concordancia fue moderado en 9 casos, bueno en 2 y muy bueno en los 2 restantes. Los mejores resultados de concordancia entre algoritmos no relacionados se obtuvieron para webPSSMX4R5/WetcatPART (k: 0,600) y webPSSMSINSI/Charge rule (k: 0,590).
Concordancia entre los diferentes algoritmos de predicción genotípica del tropismo viral en pacientes infectados con VIH-1 subtipo viral no-B (índice de kappa y grado de significación).
| geno2pheno | PSSMX4R5 | PSSMSINSI | WetcatSVM | Charge Rule | PART | C4.58a12 | C4.5 | |
| geno2pheno | X | |||||||
| PSSMX4R5 | NS (0.075) | X | ||||||
| PSSMSINSI | NS (0,243) | 0,826 (0,000) | X | |||||
| WetcatSVM | 0,586 (0,018) | 0,538 (0,029) | NS (0,142) | X | ||||
| Charge Rule | NS (0,247) | NS (0,064) | 0,590 (0,018) | NS (0,931) | X | |||
| PART | NS (0,051) | 0,600 (0,009) | NS (0,226) | 0,478 (0,025) | NS (0,226) | X | ||
| C4.58a12 | NS (0,182) | NS (0,074) | 0,448 (0,032) | NS (0,126) | 0,448 (0,032) | 0,448 (0,032) | X | |
| C4.5 | NS (0,182) | NS (0,074) | 0,448 (0,032) | NS (0,126) | 0,636 (0,006) | 0,636 (0,006) | 1,000 (0,000) | X |
En cuanto al análisis global de concordancias entre algoritmos para subtipos B (28/28, 100%) y no-B (13/28, 46,4%), se observaron diferencias significativas (p<0,001) entre los dos grupos.
DiscusiónLa aprobación del primer antagonista de CCR5 para el tratamiento de la infección por VIH, maraviroc, ha llevado a la introducción de la determinación del tropismo viral en la práctica clínica. Las herramientas genotípicas se perfilan actualmente como la alternativa a los ensayos fenotípicos para la predicción del tropismo viral antes de iniciar un tratamiento con antagonistas de CCR5.
Los diferentes algoritmos genotípicos diseñados para predecir el tropismo viral en base a la secuencia de la región V3 se han desarrollado teniendo en cuenta diferentes bases de datos de muestras genotípicas/fenotípicas pareadas en las que se han empleado distintos métodos estadísticos para identificar posiciones asociadas con un tropismo R5 o X426,27. Por tanto, las predicciones para una misma muestra pueden variar en función del método empleado. Además, la exactitud de estas herramientas en la predicción del tropismo viral podría estar también influenciada por el subtipo genético del aislado viral.
Recientemente se ha demostrado en un análisis retrospectivo de los ensayos MOTIVATE y MERIT que el ensayo fenotípico de Trofile® y algunos algoritmos genotípicos como son comparables en la predicción de respuesta a maraviroc20,21. En este contexto resulta de interés conocer el grado de concordancia entre las distintas herramientas genotípicas, así como la influencia que el subtipo viral puede tener en esta correlación.
Este trabajo analizó la concordancia de los distintos algoritmos genotípicos para la determinación del tropismo viral en un grupo de pacientes VIH+ en función del subtipo genético. Se observó una concordancia significativa entre todos los predictores genotípicos entre sí para pacientes infectados con variantes VIH-1 subtipo B. En este caso, los valores de concordancia más altos, medidos por el índice kappa, se ha encontrado para los algoritmos geno2pheno y webPSSM. Diferentes estudios han demostrado que ambos algoritmos presentan los mejores datos en términos de sensibilidad y especificidad para la detección de variantes virales X4 trópicas cuando se comparan con el ensayo fenotípico de Trofile®15,18. Además, recientemente se ha demostrado que geno2pheno y webPSSM resultan comparables a Trofile® en la predicción de respuesta a maraviroc20,21. La relación significativa obtenida en el presente estudio entre webPSSM y geno2pheno, así como el elevado valor de kappa (0,574) apoyan que ambos predictores genotípicos se comportan igual para pacientes infectados con subtipo B. Estos datos apoyados por los datos de respuesta clínica presentados recientemente20,21, apoyan que ambos predictores pueden utilizarse de forma fiable en la selección de pacientes candidatos a iniciar un tratamiento con maraviroc.
En el caso de subtipos no-B se encontró un menor grado de concordancia entre las predicciones del tropismo entre los distintos algoritmos genotípicos, siendo sólo significativa en 13 de las 28 combinaciones posibles. Aunque el número de pacientes infectados por subtipos no-B incluidos en el estudio es pequeño, estos resultados señalan una gran heterogeneidad en las predicciones de tropismo en pacientes infectados por subtipos no-B. Además, los datos disponibles hasta el momento en cuanto a la fiabilidad de las herramientas genotípicas para la determinación del tropismo en subtipos no-B muestran, en general, una peor sensibilidad para detectar variantes X4-trópicas en subtipos no-B en comparación con subtipos B (i.e. WebPSSMX4/R5: 58 vs. 89%)16,19. Otros estudios señalan, que determinadas herramientas genotípicas como geno2pheno y webPSSM pueden dar predicciones fiables para determinados subtipos no-B en concreto para el subtipo C o la forma recombinante CRF02_AG28–30. La baja sensibilidad de las herramientas genotípicas para identificar variantes X4-trópicas en muestras de pacientes infectados por subtipos no-B puede explicarse en parte, porque los algoritmos genotípicos se fundamentan en bases de datos donde de forma mayoritaria se incluyen secuencias de subtipo B. Actualmente los datos disponibles sobre la capacidad de las herramientas genotípicas para seleccionar entre pacientes respondedores y no respondedores a maraviroc en pacientes infectados con subtipos no-B son muy escasos. Por tanto, parece importante conocer el subtipo genético del paciente candidato a iniciar maraviroc.
Existen limitaciones en el presente estudio a tener en cuenta. La primera de ellas es que no se disponen de datos fenotípicos para conocer el grado de concordancia con las predicciones genotípicas ni con la evolución clínica del paciente después de la prescripción de maraviroc. Por tanto, no es posible identificar cual es la herramienta genotípica más fiable para la determinación del tropismo en la práctica clínica. En segundo lugar, podemos citar la escasa prevalencia de pacientes infectados con subtipos no B. Por último, es importante tener en cuenta la elevada proporción de pacientes naive incluidos en el estudio, en esta clase de pacientes la prevalencia de variantes X4 trópicas es menor que en pacientes pre-tratados (18-25% vs. 37-41%, respectivamente)31,32. La baja prevalencia de virus X4-trópicos en la población de estudio podría favorecer una mayor concordancia entre los predictores genotípicos si tenemos en cuenta la baja sensibilidad de estos para la detección de virus X4 trópicos.
Con estos resultados podemos concluir, que las estimaciones relativas al tropismo del VIH-1 utilizando herramientas bioinformáticas basadas en las secuencias de la región variable V3 de la envuelta viral son más homogéneas en pacientes infectados con subtipo B del VIH-1 que para los subtipos no-B, donde en general se observa un menor grado de concordancia entre algoritmos.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
Este trabajo se ha realizado con ayudas de Red de Investigación en sida (ISCIII-RETIC RD06), Fondo de Investigación Sanitaria (CP08/00214), Agencia Laín Entralgo y Fundación Investigación y Educación en Sida (FIES).

