




Tyrosinaemia type 1 (HT-1) is an autosomal recessive inherited metabolic disease affecting the fumarylacetoacetate hydrolase (FAH) gene in chromosome 15 (q23–q25) that results in altered activity of the FAH enzyme, the last enzyme implicated in the degradation pathway of the amino acid tyrosine.1 This leads to abnormal accumulation of tyrosine and its metabolites, which are cytotoxic, causing neurological, renal and liver damage. Combined treatment with a low-tyrosine diet and nitisinone (NTBC) has resulted in a greater than 90% survival rate, whereas interrupting NTBC treatment can still result in severe neurological crises that closely resemble acute porphyric attacks.2,3 Few data in the literature expose heme infusion as a treatment in paediatric patients with tyrosinaemia crisis that do not respond to initial therapy.
We report the clinical case of a twenty-five-year-old woman, the first daughter of a consanguineous Spanish couple of gypsy ethnicity. At the age of five months, she was diagnosed with HT-1. She received treatment with a low protein diet and NTBC (Orfadin; Sobi, Stockholm, Sweden), although with poor compliance. At the age of sixteen, she discontinued treatment with NTBC, missed scheduled appointments and developed obesity (maximum weight 106kg, BMI 40.3kg/m2).
In November 2022, she was admitted to the emergency department of our hospital reporting ten days of weakness, abdominal pain, urinary incontinence and lower leg pain with falls. At the time of the admission, a neurological examination revealed flaccid paralysis and bilateral areflexia of the four extremities. Blood analysis showed leukocytosis without elevation of acute phase reactants, thrombocytopenia, altered coagulation, mild hyponatraemia, hypokalaemia, and metabolic alkalosis with a normal liver and renal function. In the urine, and later in the blood culture, an extended spectrum beta-lactamase Escherichia coli was isolated. In light of these findings, the patient was diagnosed with acute decompensation of HT-1 in the context of a urinary sepsis. NTBC at a dose of 1mg/kg/day was initiated, in addition to vitamin K and ceftriaxone.
Eight hours later, in the context of a fever peak of 38°C, she suffered from two consecutive episodes of decreased level of consciousness with myoclonic movements, the last one without recovery of the level of consciousness. The patient was then transferred to the Intensive Care Unit (ICU) for mechanical ventilation. She continued antiepileptic treatment without repeating any seizure crisis.
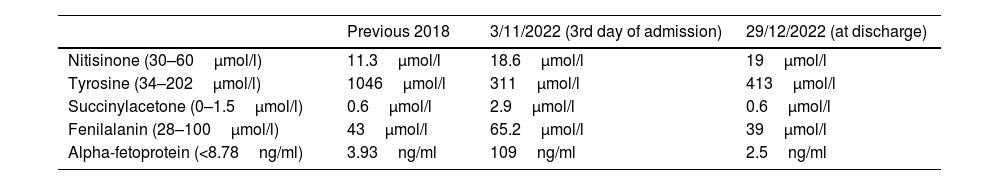
Following Canadian consensus group review and recommendations for HT-1,4 specific nutrition without tyrosine or phenylalanine was progressively introduced through a nasogastric tube (ProZero, Vitaflo) with the addition of a powdered specific protein module for tyrosinaemia (Tyr sphere, Vitaflo) under the care of a registered dietitian. The NTBC dose was maintained at 1mg/kg/day. A complete metabolic analysis was performed (Table 1), highlighting high levels of succinylacetone and alpha-fetoprotein with normal liver function. In the following days, there was a decline in peripheral neuropathy with generalised areflexia and an electromyogram identified axonal motor polyradiculoneuropathy. In the face of the lack of improvement, the NTBC dose was increased to 1.5mg/kg/day.
Main laboratory results.
| Previous 2018 | 3/11/2022 (3rd day of admission) | 29/12/2022 (at discharge) | |
|---|---|---|---|
| Nitisinone (30–60μmol/l) | 11.3μmol/l | 18.6μmol/l | 19μmol/l |
| Tyrosine (34–202μmol/l) | 1046μmol/l | 311μmol/l | 413μmol/l |
| Succinylacetone (0–1.5μmol/l) | 0.6μmol/l | 2.9μmol/l | 0.6μmol/l |
| Fenilalanin (28–100μmol/l) | 43μmol/l | 65.2μmol/l | 39μmol/l |
| Alpha-fetoprotein (<8.78ng/ml) | 3.93ng/ml | 109ng/ml | 2.5ng/ml |
On the basis of previous literature in paediatric patients,5,6 as severe neurological crises in HT-1 closely resemble acute attacks seen in the acute porphyrias, human heme infusion (NORMOSANG 25mg/ml, Recordati Rare Diseases, France) was administered at doses of 3mg/kg for four days. An intensive rehabilitation programme was also initiated. Six days after receiving the heme treatment, an improvement in motor skills was observed in all four limbs and the abdominal pain subsided. Laboratory tests returned to normal and delta-aminolevulinic acid (ALA) urine levels were in normal range: 4.1μmol/mol creatinine (normal<5.0). Unfortunately, ALA levels prior to heme treatment were not available. After 57 days of ICU admission, it was possible to transfer the patient to a medical ward.
Due to improved clinical and nutritional status, the NTBC dose was reduced to 1mg/kg/day and it was possible to remove the nasogastric tube. A low protein diet plus specific oral nutritional supplements were prescribed with good compliance. Blood analysis at discharge revealed normalisation of succinylacetone and alpha-fetoprotein levels. Nevertheless, as NTBC levels were below range, we increased the dose again to 1.5mg/kg/day. The patient was transferred to a neurorehabilitation hospital to undergo physical and nutritional therapy.
Nowadays there is convincing evidence that ALA is the cause of pain in acute porphyria attacks. The benefit of heme therapy for neurological crises or acute porphyria attacks is the direct ALA-lowering effect due to the inhibition of hepatic delta-aminolevulinic acid synthase-1 (ALAD). NTBC indirectly lowers ALA by lowering the production and accumulation of succinylacetone, and reducing succinylacetone levels leads to recovery of ALAD enzyme activity.7
Rochus A. et al. describe a paediatric patient with HT-1 with several acute porphyric attacks due to noncompliance with NTBC treatment. It was impossible to restart NTBC orally due to nausea and heme therapy was needed to quickly resolve the acute decompensation.5 Furthermore, there is a report that found a significant correlation between BMI and adherence to NTBC treatment; thus, patients with a higher BMI comply less with treatment,8 than the patient we present.
One limitation of this case is that we did not obtain ALA levels prior to starting heme treatment. Neither cannot confirm that the improvement in peripheral neuropathy is mainly attributed to intravenous heme therapy, or more probably to the combination of heme therapy with the previous dose increase of NTBC to 1.5mg/kg/day. Moreover, we did not study whether the patient had the variant in the FAH gene.
To the best of our knowledge, this is the first report of an adult patient with severe decompensation of HT-1 not responding to NTBC, which improved significantly one week after the introduction of heme infusion. We suggest that in adult patients with an acute decompensation of HT-1 resembling a porphyric attack, heme therapy could be an add-on treatment to NTBC in order to improve clinical prognosis. Moreover, an intensive rehabilitation programme and systematic positive reinforcement at medical visits should be mandatory in these cases.