



Parkinson’s disease (PD) is a neurodegenerative disorder that affects more than 7 million people worldwide. Its aetiology is unknown, although the hypothesis of a genetic susceptibility to environmental agents is accepted. These environmental agents include fungi, bacteria, and viruses. Three microorganisms are directly associated with a significantly increased risk of developing Parkinson’s disease: the fungal genus Malassezia, the bacterium Helicobacter pylori, and the hepatitis C virus. If the host is vulnerable due to genetic susceptibility or immune weakness, these microorganisms can access and infect the nervous system, causing chronic neuroinflammation with neurodegeneration. Other microorganisms show an epidemiological association with the disease, including the influenza type A, Japanese encephalitis type B, St Louis, and West Nile viruses. These viruses can affect the nervous system, causing encephalitis, which can result in parkinsonism. This article reviews the role of all these microorganisms in Parkinson’s disease.
La enfermedad de Parkinson es un trastorno neurológico degenerativo que afecta a más de siete millones de personas en todo el mundo. Se desconoce su etiología, aunque se acepta que existe una susceptibilidad genética a agentes ambientales. Estos agentes ambientales incluyen hongos, bacterias y virus. Tres microorganismos están directamente relacionados con un mayor riesgo estadístico de padecer enfermedad de Parkinson: el género de hongos Malassezia, la bacteria Helicobacter pylori y el virus de la hepatitis C. Estos microorganismos, si el huésped es vulnerable por susceptibilidad genética o debilidad inmunológica, pueden acceder al sistema nervioso, infectarlo y causar neuroinflamación crónica con neurodegeneración. Otros microorganismos se relacionan desde una vertiente epidemiológica con la enfermedad, destacando los virus influenza tipo A, de la encefalitis japonesa tipo B, de San Luis y del Nilo Occidental. Estos virus pueden afectar al sistema nervioso causando encefalitis, cuya consecuencia puede ser un parkinsonismo. En este artículo se hace una revisión de los mencionados agentes infecciosos en la enfermedad de Parkinson.
Parkinson’s disease (PD) is the second most frequent neurodegenerative disease, affecting more than 1% of individuals older than 65 years. The pathological hallmark of PD is degeneration and death of dopaminergic neurons in the substantia nigra pars compacta, a central nervous system (CNS) region playing a key role in motor control. PD is diagnosed based on the classic triad of motor symptoms: bradykinesia, rigidity, and resting tremor.1,2 In terms of anatomical pathology, PD is classified as an α-synucleinopathy, that is a disorder caused by the abnormal accumulation of α-synuclein aggregates in neurons.3–6 Aggregation of α-synuclein leads to the formation of Lewy bodies and consequently to the loss of dopaminergic neurons in the substantia nigra.5–7 PD is a multifactorial disorder whose pathogenesis involves such factors as neuroinflammation, oxidative stress, infection, and genetic factors. In this review, we focus on infection and neuroinflammation, 2 factors that are receiving increasing attention in the study of PD pathogenesis.8–10
Infection as a risk factor for Parkinson’s diseaseRecent meta-analyses suggest that at least 3 microorganisms are directly associated with a significant increase in the risk of developing PD. These microorganisms are fungi from the genus Malassezia, the bacterium Helicobacter pylori, and hepatitis C virus (HCV).11,12 From an epidemiological viewpoint, other microorganisms have been associated with PD, particularly viruses causing encephalitis associated with parkinsonism, such as influenza A virus, Japanese encephalitis virus, St. Louis encephalitis virus, and West Nile virus.13
Although infections caused by Malassezia spp, H pylori, and HCV are very frequent in humans, only a minority will develop PD. Why should this be? It is widely accepted that certain microorganisms may promote the development of PD only in highly susceptible individuals, and when certain pathological conditions are met. Firstly, the pathogen must be neurotropic (ie, able to invade the CNS). Secondly, it must cause acute cell death and a strong immune response in the brain, triggering a cytokine storm. Thirdly, once the cytokine storm resolves, the patient must present long-term sequelae. In other words, patients would develop chronic neuroinflammation as a result of persistent activation of microglia and astrocytes (the main immune cells in the CNS), frequently facilitated by a deficient immune response, in which CD4+ T cells proliferate in the presence of the pathogenic agent, but their immune response (based on the release of cytokines) is weak.14 Other authors have proposed the so-called dual-hit hypothesis, according to which a first attack would sensitise the brain to a subsequent, probably environmental attack, which would otherwise not be pathogenic, and which would activate the microglia.15–17 In any case, a comprehensive study of the mechanisms by which infection increases the risk of PD may deepen our understanding of the pathophysiology of the disease.
Neuroinflammation as a pathological process in Parkinson’s diseaseChronic neuroinflammation is receiving increasing attention as a pathological process in PD.18,19 Patients with PD are known to present chronic activation of microglia and astrocytes, manifesting as an increase in the levels of such cytokines as interleukins (IL-1β, IL-2, IL-4, IL-6), tumour necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ). This results in more marked inflammation and lymphocytic infiltration, and presence of blood-borne macrophages in the CNS.20,21 Despite the presence of lymphocytic infiltration, CD4+ T cells present a deficient response, as mentioned previously, probably due to IFN-γ overactivation.14 Presence of chronic inflammation is reflected by elevated blood C-reactive protein levels at any stage of PD.22 Furthermore, levels above 0.69 mg/L are associated with more severe motor impairment, with very high levels being associated with poor vital prognosis.23,24 The following sections focus on the pathogens that have been found to be associated with chronic neuroinflammation causing secondary neurodegeneration.
Malassezia spp fungiMalassezia spp proliferate on the skin and in the gastrointestinal tract of patients with Parkinson’s diseaseMalassezia spp fungi are naturally found on the human skin, particularly in hair follicles. Seven species have been identified on the human skin: Malassezia globosa, Malassezia furfur, Malassezia slooffiae, Malassezia sympodialis, Malassezia restricta, Malassezia obtusa, and Malassezia japonicа. Although cutaneous symptoms are very frequent in PD, few studies have focused on skin alterations or the skin microflora in these patients. Interestingly, the concept of the skin being a reflection of PD has existed for more than a century.25 The importance of the skin in PD was demonstrated over a decade ago, with studies showing the presence of α-synuclein deposits in cutaneous autonomic nerves.26–28
Most, if not all, patients with PD present dermatological diseases, such as hyperhidrosis, folliculitis, or seborrheic dermatitis, which promote fungal growth. More specifically, seborrheic dermatitis affects over 60% of patients with PD, compared to 3% of the general population.29,30 Some authors reject the idea of a causal relationship between PD, skin inflammation, and Malassezia spp, and suggest that fungal infections in fact constitute a comorbidity.31,32Malassezia spp are opportunistic, lipophilic pathogens that proliferate when sebum accumulates, and seborrheic dermatitis and folliculitis are associated with excessive sebum secretion and alterations in sebum composition.33–35
However, there is evidence of a more direct, non-opportunistic relationship between Malassezia spp and PD. One cause of alterations of sebum composition is ageing, which is also the main risk factor for PD.36 Skin inflammation usually appears prior to diagnosis of PD. Furthermore, Malassezia spp are associated with genetic forms of parkinsonism, since the genetic polymorphisms associated with PD (located in such genes as LRRK2, GBA, PINK1, SPG11, and SNCA) increase the availability of lipids, particularly saturated long-chain fatty acids and gangliosides, within human cells, promoting a favourable environment for Malassezia species.35,37
Malassezia spp, and especially M restricta, are also found in the gastrointestinal tract. They are commensal microorganisms normally present in the gut microbiota, but they proliferate and become pathogenic when the microbiota is altered. This occurs in gastrointestinal disorders, which affect over 80% of patients with PD, and such inflammatory intestinal disorders as colitis, which are also very prevalent among patients with PD.38 As is the case with skin inflammation, gastrointestinal alterations may present several years before PD is diagnosed. Enteric inflammation damages the gastrointestinal mucosa, facilitating the invasion of pathogens via the transneuronal or haematogenous routes.39,40 Braak et al.41 even postulate that pathological processes in PD originate in the gastrointestinal tract, extending to the CNS via the vagus nerve. Interestingly, vagotomy is associated with lower risk of PD.42,43
Chronic intestinal inflammation is accompanied by a powerful neuroinflammatory response in the intestinal wall, with production of cytokines and IgE targeting Malassezia. This phenomenon is more marked in individuals with a genetic predisposition associated with a polymorphism in the CARD9 gene, which synthesises the CARD9 protein, a critical factor in the antifungal immune response.44 Interestingly, CARD9 deficiency also promotes fungal invasion of the CNS.45 In summary, we cannot rule out the pathogenic role of enteric Malassezia fungi in patients with PD, especially in patients who also present a genetic predisposition due to CARD9 polymorphisms.
Malassezia spp consume levodopaMalassezia fungi produce melanin from levodopa, a dopamine precursor and the main pharmacological treatment for PD.46Malassezia fungi may be defined as levodopa-tropic, and grow more easily in the presence of levodopa. Levodopa also promotes the growth of hyphae, associated with more aggressive infections. These facts suggest that Malassezia spp may be more invasive in regions rich in levodopa, such as the substantia nigra, which contains dopaminergic neurons.47 Lastly, levodopa use in patients with PD may promote the growth of Malassezia fungi, particularly on the skin and in the gastrointestinal tract, although this hypothesis has not been demonstrated.48
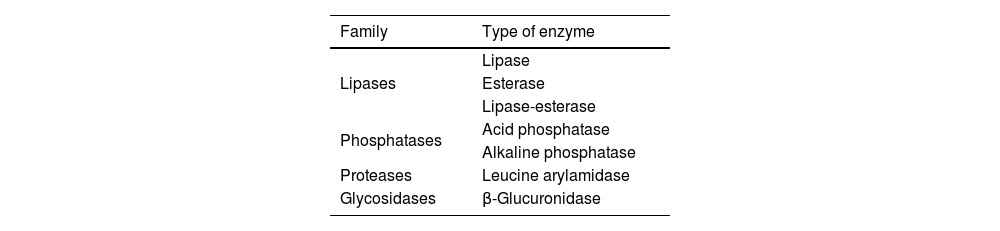
Malassezia spp release proinflammatory and antigenic factorsInflammation caused by Malassezia fungi is exacerbated and becomes chronic due to the nature of their enzymes.29,35 Fungi are able to consume cellular lipids, obtaining free fatty acids through the action of lipases and phosphatases. Their main energy source are saturated long-chain fatty acids, which are abundant in the sebum and human diet, and whose concentration varies with age, immune status, and neurodegeneration. Fungal lipases and phosphatases may trigger inflammatory responses by releasing oleic and arachidonic acids from the sebum lipids. These acids originate free fatty acids and also such proinflammatory cytokines as IL-1β, IL-2, IL-5, and IL-6.
Such other fungal enzymes as β-glucuronidase and leucine arylamidase may act as antigens in genetically predisposed individuals, and can modulate the host’s immune response. They promote inflammation and weaken the T-cell response in tissues colonised by the fungus.29,49 The most important fungal enzymes are listed in Table 1. If Malassezia fungi were to invade the CNS, infection would activate the microglia and increase tissue levels of inflammatory factors of microglial origin, such as TNF-α, IFN-γ, and IL.
Malassezia spp may invade the central nervous systemAlthough presence of Malassezia spp has not been confirmed in the substantia nigra, the fungus is found in the CNS of patients with PD, as demonstrated in a study by Pisa et al.50 The authors detected Malassezia spp in the mesencephalon, brainstem, and motor cortex, although they did not perform a detailed study of nerve nuclei. In their study, only fungi from the genera Malassezia (M globosa and M restricta) and Cladosporium (very common fungi) were detected in the CNS of all patients.
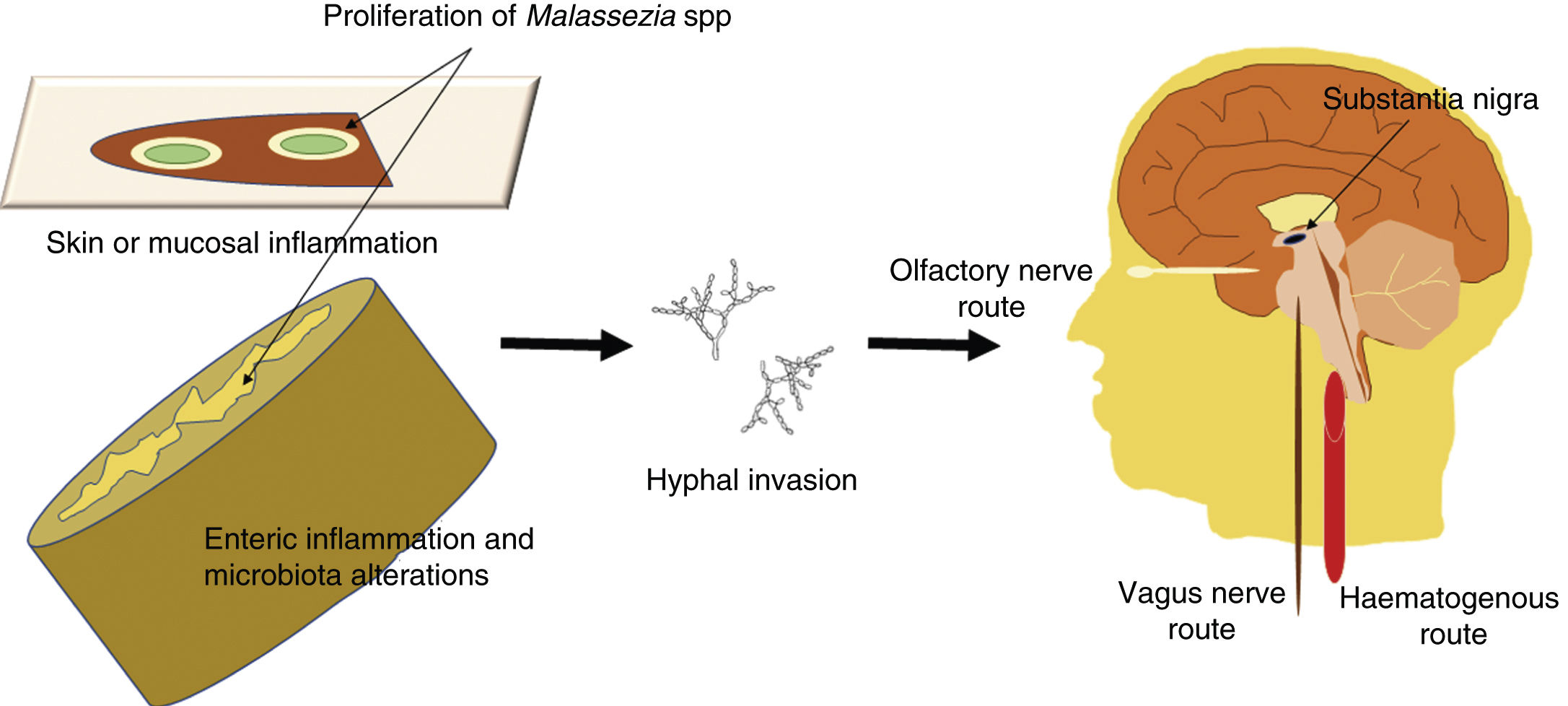
Malassezia infection is known to be associated with increased risk of PD35,50; although infection may simply constitute a comorbidity, a direct role in the disease cannot be ruled out. Given that these fungi are opportunistic microorganisms, we may hypothesise that Malassezia spp may exploit the host’s vulnerabilities to reach the CNS, probably via the haematogenous route or such peripheral nerves as the vagus or the olfactory nerves. Opportunistic infection may occur in the event of a genetic predisposition, a weakened immune system, gastrointestinal tract inflammation with altered microbiota, or damage to the skin or mucosal barriers. A schematic representation of the invasion routes of Malassezia spp is shown in Fig. 1. Malassezia spp. are commensal microorganisms frequently found on the human skin and in the gut microbiota, but they may become pathogenic if they proliferate and develop hyphae.51 The mechanism by which fungi invade the CNS is yet to be understood.52
 invasion by Malassezia spp. These fungi are opportunistic microorganisms that exploit the host’s vulnerabilities to invade the CNS, damaging neurons and causing neuroinflammation. Opportunistic infection may occur in the event of a genetic predisposition, a weakened immune system, gastrointestinal inflammation with altered microbiota, or damage to the skin or mucosal barriers. Malassezia spp are most invasive when they grow hyphae; hyphal fungi may enter the CNS by the transneuronal route via the olfactory or the vagus nerves, or by the haematogenous route. They may settle in the substantia nigra, an area rich in dopaminergic neurons, as these fungi consume levodopa and would therefore present a preference for levodopa-rich regions.")
Schematic representation of the possible routes of central nervous system (CNS) invasion by Malassezia spp. These fungi are opportunistic microorganisms that exploit the host’s vulnerabilities to invade the CNS, damaging neurons and causing neuroinflammation. Opportunistic infection may occur in the event of a genetic predisposition, a weakened immune system, gastrointestinal inflammation with altered microbiota, or damage to the skin or mucosal barriers. Malassezia spp are most invasive when they grow hyphae; hyphal fungi may enter the CNS by the transneuronal route via the olfactory or the vagus nerves, or by the haematogenous route. They may settle in the substantia nigra, an area rich in dopaminergic neurons, as these fungi consume levodopa and would therefore present a preference for levodopa-rich regions.
Hpylori is found in half of the population; in patients with PD, however, the risk of Hpylori infection is 3 times higher than in the general population,53 resulting in a prevalence of 35%-70% in patients with PD.54 Furthermore, individuals with Hpylori infection present an increase of 1.5 to 2 times in the risk of developing PD.55
Gastric inflammation caused by Hpylori in turn causes enteric inflammation, which alters the gut microbiota. An altered microbiota produces inflammatory factors, alters the host immune response, and promotes α-synuclein aggregation in the vagus nerve; these processes are associated with the pathogenesis of PD.56–58 As occurs with Malassezia spp, enteric inflammation damages the intestinal barrier and promotes invasion by pathogens via the transneuronal or haematogenous routes.39,40
This abnormal interaction of the gut-brain axis in patients with PD is widely known. Certain genetic factors are known to underlie this abnormal interaction, increasing an individual’s susceptibility to developing PD. These genetic factors include overexpression of the CARD15 gene (associated with colitis and Crohn disease),59 DNA overmethylation in the SNCA gene, which generates abnormal α-synuclein with greater capacity for aggregation,57 and TREM2 gene variants associated with increased risk of PD.60
Helicobacter pylori decreases the effectiveness of levodopaPatients with PD testing positive for Hpylori display a poorer motor response to levodopa,61–63 as the bacterium decreases the effectiveness of the drug. In fact, Hpylori eradication increases plasma levels of levodopa and improves motor response in patients with PD.64–66 The bacterium and the drug are thought to interact at 3 different levels. Firstly, Hpylori alters the gastric pH level, which results in decreased absorption of levodopa.67 Secondly, Hpylori consumes levodopa, at least in vitro, which in turn decreases the drug’s bioavailability.68 Lastly, the bacterium binds to the drug by means of adhesins located on the bacterial membrane, inducing enteric inflammation and consequently decreasing the intestinal absorption of levodopa.67,68
Helicobacter pylori produces neurotoxic and antigenic factorsAnother potential explanation for the increased risk of PD associated with Hpylori infection is the production of toxins and antigens with detrimental effects on neurons. Hpylori releases such toxins as VacA and CagA, which have neurotoxic effects on dopaminergic neurons. In fact, immunoblot analyses detect CagA in the serum of patients with PD testing positive for Hpylori, which suggests that this toxin may affect the CNS.69Hpylori contains cholesterol-α-glucosyltransferase, which glycosylates membrane cholesterol, resulting in 3 glycosylated derivatives. These derivatives exert a neurotoxic effect on dopaminergic neurons and promote α-synuclein aggregation in the vagus nerve at the level of the stomach.70–72 As mentioned previously, it has been hypothesised that the pathological processes of PD extend to the CNS through the vagus nerve.41
Hpylori produces fucosylated proteins, which have antigenic properties as they mimic human cell surface glycoconjugates.73 This antigenic effect generates autoantibodies that may react against dopaminergic neurons or trigger a cytokine storm in the brain, exacerbating neurodegeneration.71,74 We should also mention that Hpylori has not been detected in the substantia nigra, brain tissue, or bloodstream of patients with PD.74
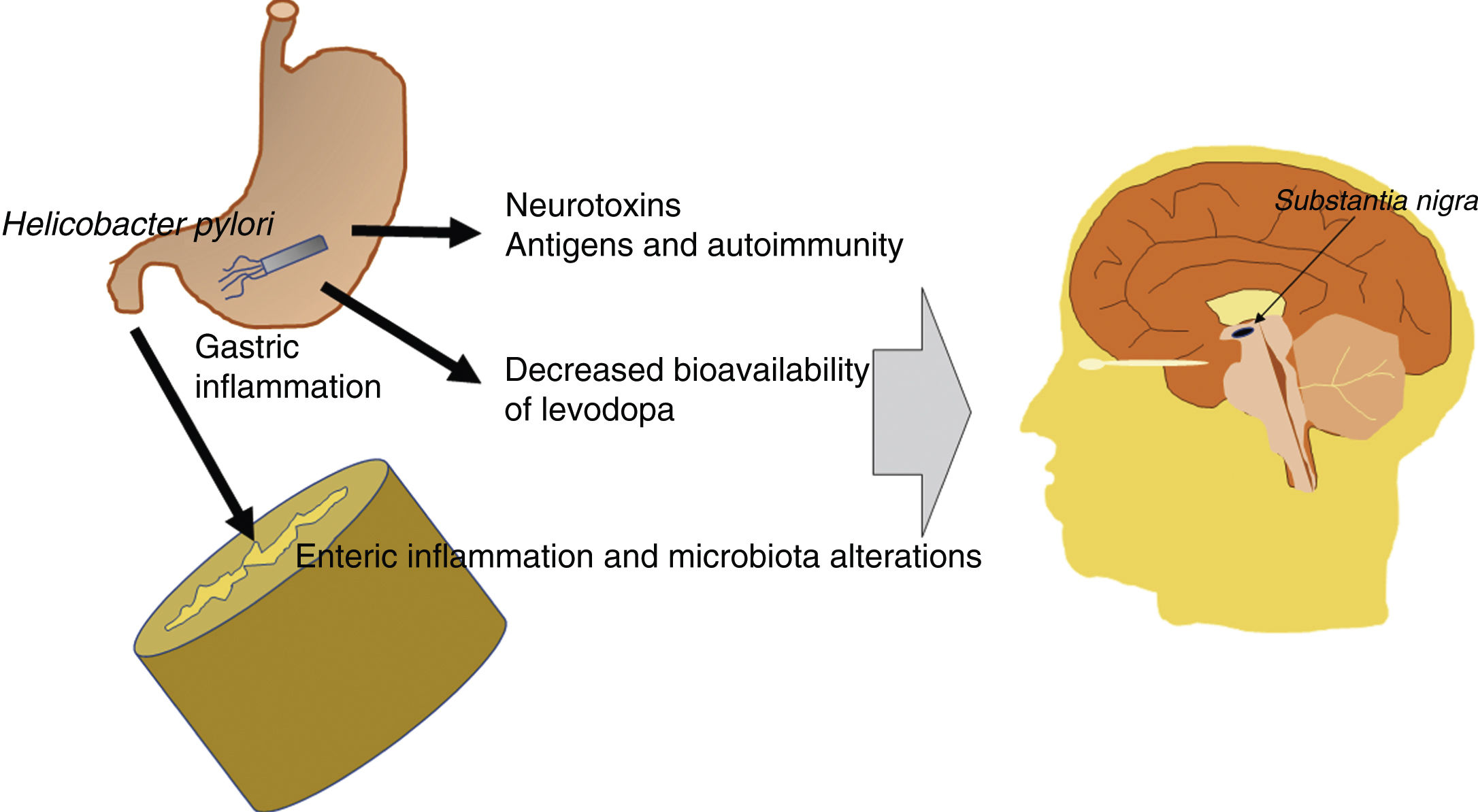
In summary, the pathogenic effects of Hpylori are either indirect or linked to the bioavailability of levodopa. The bacterium produces neurotoxic and antigenic factors with detrimental effects for dopaminergic neurons, and induces gastrointestinal inflammation, which alters the gut microbiota, making it pathogenic. This has a negative impact on individuals genetically predisposed to PD. Furthermore, the therapeutic effectiveness of levodopa decreases in patients with Hpylori infection. Fig. 2 presents a schematic representation of the possible pathogenic mechanisms of Hpylori in PD.

Schematic representation of the potential pathogenic mechanisms of Helicobacter pylori in Parkinson’s disease. Hpylori produces neurotoxic and antigenic factors, which damage dopaminergic neurons in the substantia nigra. It also induces gastrointestinal inflammation, which alters the gut microbiota, making it pathogenic. Levodopa presents reduced bioavailability and therapeutic effectiveness in patients with Parkinson’s disease and Hpylori infection.
Hepatitis C virus infection is associated with an increase of 1.5-2 times in the risk of developing PD,75–78 although this increase is eliminated after administration of effective antiviral therapy.79,80 Some individuals seem to be particularly susceptible; in these patients, HCV can cross the blood-brain barrier and trigger a cytokine storm and neuroinflammation in the brain, damaging dopaminergic neurons in the substantia nigra.75 These individuals present a specific receptor profile, including such membrane proteins as CD81, claudin-1, LDLR, and occludin. These receptors are mainly expressed in endothelial cells of brain microvessels, allowing massive entry of the virus into the human brain.75,81
Viruses with an epidemiological association with Parkinson’s diseaseInfluenza A virusMany subtypes of influenza A viruses are neurotropic; therefore, in addition to respiratory symptoms, they may also cause CNS alterations,82,83 including encephalitis and neuroinflammation, which may lead to neurodegeneration in the long term.83,84 Influenza A virus infection is known to cause postencephalitic parkinsonism, as demonstrated by the increase in PD incidence after such influenza outbreaks as the 1918 pandemic, caused by the H1N1 strain, which resulted in over 30 million deaths.85,86 Furthermore, people born around 1918 presented 2-3 times more risk of developing PD than individuals born after 1924.87,88 From an experimental viewpoint, the H1N1 strain is known to infect dopaminergic neurons in the substantia nigra and to activate pathological cellular mechanisms underlying PD, such as mitochondrial dysfunction, endoplasmic reticulum stress, and autophagy deficiency.89,90
Experimental studies have shown that other subtypes of influenza A virus, such as H5N3, H3N2, and H7N7 (some of which caused such outbreaks as the 1968 pandemic, caused by the H3N2 strain, with over a million deaths), trigger cytokine storms in the brain and cause dopaminergic damage.91–94 Although these strains do not cause encephalitis, chronic neuroinflammation secondary to the cytokine storm may cause parkinsonism in the long term.12 In fact, such coronaviruses as SARS-CoV-2, which caused the COVID-19 pandemic, trigger a strong cytokine storm in the human brain and alter mitochondrial function and cell autophagy.95–97 This could translate into a greater risk of PD, although this hypothesis will only be confirmed with time.
Other viruses causing postencephalitic parkinsonismAs mentioned previously, other viruses that cause encephalitis are also associated with parkinsonism. These include Japanese encephalitis virus, St. Louis encephalitis virus, and West Nile virus.13 The type of encephalitis caused by these viruses may be associated with atypical parkinsonism, presenting with the classic symptoms of PD, but which may not respond to levodopa and is not associated with presence of Lewy bodies.98–102
ConclusionsAlthough the aetiology of PD is unknown, the hypothesis of a genetic predisposition to certain environmental triggers is widely accepted. Environmental factors include infection with certain fungi, bacteria, and viruses. Three microorganisms have been directly associated with a statistically significant increase in the risk of developing PD: Malassezia spp, H pylori, and HCV. In individuals with a genetic predisposition or immune deficiency, these pathogens can invade the CNS, causing chronic neuroinflammation and neurodegeneration. Other pathogens associated with PD from an epidemiological viewpoint are influenza A virus, Japanese encephalitis virus, St. Louis encephalitis virus, and West Nile virus. These viruses can invade the CNS, causing encephalitis, which may ultimately lead to the development of parkinsonism.
FundingThis study has received no funding.
Conflicts of interestThe author has no conflicts of interest to declare.
The author wishes to thank Fernando Rodríguez de Fonseca (Institute of Biomedical Research of Malaga), Antonio Córdoba Fernández, and Ángel Martín de Pablos (University of Seville) for their invaluable help and friendship.
