







La enfermedad pulmonar intersticial en el lupus es una entidad que se presenta con poca frecuencia y en la mayoría de los casos tiende a ser de progresión lenta. A pesar de esto, se desconoce en gran medida el enfoque terapéutico de los casos moderados a severos, debido a que la mayor parte de la evidencia proviene de reportes de caso y muchos de ellos fueron anteriores al advenimiento de los nuevos tratamientos para el lupus que se conocen hoy en día. Adicionalmente, se ha avanzado poco en entender su fisiopatología, los conceptos actuales provienen de otras enfermedades del tejido conectivo como la esclerosis sistémica que en ocasiones son agrupadas dentro del grupo de neumonías intersticiales con características autoinmunes. Esto de cierta forma ha sido un obstáculo para la investigación en este campo, sin que se haya logrado un enfoque diagnóstico y terapéutico unificado. Por ello, se realiza una búsqueda avanzada con el objetivo de tener la mayor evidencia disponible hasta la fecha en cuanto a los métodos diagnósticos y las terapias emergentes, ofreciendo al clínico una visión práctica para lograr su abordaje integral.
Interstitial lung disease in lupus is an entity that occurs infrequently and tends to progress slowly in most cases. Despite this, the therapeutic approach for moderate to severe cases is largely unknown because most of the evidence comes from case reports, many of which predate the advent of today's known treatments for lupus. Additionally, little progress has been made in understanding its pathophysiology and current concepts come from other connective tissue diseases such as systemic sclerosis or are grouped within the group of interstitial pneumonias with autoimmune characteristics. This, to an extent, has been an obstacle for research in this field, and to date there is no unified diagnostic and therapeutic approach. Therefore we conducted a state of the art search of the best evidence available to date, in terms of diagnostic methods and emerging therapies, to offer the clinician a practical vision for a comprehensive approach.
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune sistémica de severidad variable con tendencia a presentar brotes en el trascurso de su evolución. Las alteraciones inmunológicas, particularmente la producción de diversos anticuerpos antinucleares, son una de sus características determinantes. En su fisiopatología intervienen tanto el sistema inmune innato como el adaptativo, así como la interacción entre genes con factores ambientales que causan alteraciones inmunológicas sostenidas contra ácidos nucleicos autólogos1. En algún momento durante el curso de la enfermedad, la mayoría de los pacientes con LES muestran signos de compromiso pulmonar (ya sea del parénquima o de su vasculatura), la pleura o el diafragma2,3. El dolor pleurítico, la tos o la disnea pueden ser los primeros indicios del compromiso pulmonar o ser propios del comienzo del LES4.
A pesar de que la enfermedad pulmonar intersticial (EPI) es una manifestación común en otras enfermedades del tejido conectivo, en el LES es la excepción, siendo un hallazgo inusual que compromete solo del 1% al 15% de los pacientes5. Si bien la EPI en el LES rara vez es grave y la progresión clínica suele ser lenta, dos tercios de los pacientes mostrarán anomalías asintomáticas en las pruebas de función pulmonar y un tercio va a experimentar cambios compatibles de EPI en la tomografía computarizada de tórax de alta resolución (TACAR)6,7. La prevalencia reportada del compromiso intersticial varía entre el 3% y el 8%, observándose un aumento mientras más tiempo de evolución tenga la enfermedad8. La EPI tiende a ser más común en adultos mayores de sexo masculino con una presentación tardía del LES9.
Al igual que otras enfermedades autoinmunes, la EPI en el LES puede presentar los mismos hallazgos histopatológicos de las diversas neumonías intersticiales idiopáticas como la neumonía intersticial no específica (NINE), la neumonía intersticial usual (NIU), la neumonía organizada (NO) y la neumonía intersticial linfocítica (NIL). La NINE es el patrón que se observa más comúnmente en el LES, mientras la NIU se presenta de forma infrecuente10–12. La gravedad de la enfermedad pulmonar intersticial no se correlaciona con los marcadores serológicos específicos del lupus, como los anticuerpos anti-dsDNA y el consumo del complemento. Sin embargo, se ha informado una asociación débil con anticuerpos anti-SSA/Ro13.
MetodologíaSe llevó a cabo una revisión narrativa no sistemática de la literatura en idiomas inglés y español, de acuerdo con el objetivo de tener la información más representativa disponible para los artículos referenciados en bases de datos primarias como Pubmed, Embase y Google Scholar. Los términos medical subject headings (MESH) utilizados fueron: «Enfermedad pulmonar intersticial», «Lupus eritematoso sistémico», «Enfermedades autoinmunes» y «Manifestaciones pulmonares del lupus», los cuales se combinaron utilizando operadores booleanos (AND, OR). En la figura 1 se presenta un diagrama de flujo que detalla la estrategia de búsqueda.
Fisiopatología
Se tiene poco conocimiento de la fisiopatología de la EPI en el LES, pero probablemente sea el resultado de una respuesta inflamatoria aberrante debido al desequilibrio de citocinas proinflamatorias y antiinflamatorias14. Aproximadamente el 15% de los pacientes con EPI tienen una enfermedad del tejido conectivo subyacente15. Es de destacar que casi todas las enfermedades del tejido conectivo afectan a los pulmones y producen cambios patológicos, aunque los síntomas clínicos en ocasiones pueden estar ausentes. Se sabe que tanto las células T CD4+como las células B autorreactivas producen autoanticuerpos patógenos. Varios autoanticuerpos son identificables en las diferentes enfermedades autoinmunes y en las vasculitis, pero son de poca utilidad para el diagnóstico y para la evaluación de la progresión de la EPI16.
Un estudio demostró que el ligando de quimiocinas 1 (CXCL1) y su receptor CXCR2 pueden estar involucrados en el desarrollo de neumonía intersticial con características autoinmunes (NICA). CXCL1, a través de CXCR2, actúa para reclutar neutrófilos, por lo que presumiblemente ejerce un efecto inflamatorio. Los niveles de CXCL1 estaban elevados en el plasma de pacientes con NICA y se asociaron con exacerbaciones. Además, CXCR2 estaba regulado al alza en los leucocitos y las células endoteliales de los pulmones de pacientes con NICA, en comparación con los pacientes con fibrosis pulmonar idiopática17.
También se ha descrito que los niveles iniciales de los marcadores de fibrosis, Krebs von den Lungen-6 (KL-6), y la proteína A del surfactante aumentan en los pacientes con NICA que progresan, correlacionándose negativamente con los resultados de las pruebas de función pulmonar18. Cabe resaltar que estas últimas alteraciones fisiopatológicas no se han confirmado en pacientes con LES de forma exclusiva, por lo que se requieren investigaciones adicionales para confirmar su participación en la progresión de la enfermedad pulmonar intersticial en este contexto.
Presentación clínica y marcadores serológicosLas manifestaciones clínicas de la EPI crónica asociada con LES son la tos crónica no productiva, la disnea de esfuerzo persistente y el dolor pleurítico, aunque la mayoría de los pacientes tienden a ser asintomáticos19. Al examen físico se puede observar cianosis central, estertores bibasales e hipocratismo digital, aunque este último hallazgo es menos común en el LES que en otras enfermedades pulmonares intersticiales idiopáticas20.
Los pacientes con características similares a la esclerodermia tienen un mayor riesgo de desarrollar EPI, como se ha demostrado por la asociación de esclerodactilia, capilares anormales del pliegue ungueal, anticuerpos anti-RNP y fenómeno de Raynaud. Esto sugiere que la presencia de un síndrome de superposición o una enfermedad del tejido conectivo con características mixtas de esclerodermia se relacionan con la aparición de EPI en el LES21. Los altos niveles de proteína C reactiva, la hipocomplementemia y la presencia de crioglobulinas en suero también se han asociado con su desarrollo22. Es importante evaluar otros marcadores serológicos, como anticuerpos contra antígenos nucleares extraíbles, factor reumatoide, anticuerpos antisintetasa y creatina quinasa para descartar síndromes de superposición. Las pruebas que indican una mayor actividad de la enfermedad, como la proteína C reactiva o la hipocomplementemia, se han asociado con la progresión de la EPI, pero no son útiles para su diagnóstico23.
Pruebas de función pulmonar y hallazgos histopatológicosLas pruebas de función pulmonar (PFP) se obtienen para evaluar el patrón y la gravedad de la insuficiencia respiratoria. El hallazgo de las PFP más comunes en la EPI asociada con LES es una prueba de difusión de monóxido de carbono (DLCO) disminuida; otros hallazgos pueden incluir un patrón restrictivo en la espirometría y desaturación de oxígeno durante la caminata de 6 min24,25. Una DLCO disminuida, con volúmenes pulmonares normales, puede indicar la posibilidad de una EPI en estadio temprano, aunque se debe descartar la posibilidad de una enfermedad vascular pulmonar. Los estudios histopatológicos han reportado la presencia de linfocitos e infiltrados intersticiales y peribronquiolares mononucleares en biopsias tomadas de pacientes con LES que tenían el patrón de NINE26. La fibrosis intersticial puede estar presente junto con los depósitos de IgG, IgM, C1q y C3 dentro de los tabiques alveolares27.
ImágenesLas radiografías de tórax pueden mostrar alguna evidencia de EPI, pero la TACAR es el estándar de oro para determinar si hay compromiso intersticial, definir el patrón radiológico y su correlación histopatológica específica28. Un estudio transversal de 34 pacientes con LES realizado por Fenlon et al. correlacionó los hallazgos de la TACAR, con las características clínicas, las radiografías de tórax y las PFP. El 70% mostró anomalías en la TACAR, el 24% tenía radiografías de tórax anormales, el 41% tenía PFP anormales y se consideró que el 33% tenía EPI en la TACAR. No se encontró correlación entre la TACAR anormal y los síntomas clínicos, la actividad de la enfermedad, los antecedentes de tabaquismo y el tratamiento. Los hallazgos de las PFP no se correlacionaron con la severidad de la EPI o la presencia de bronquiectasias en la TACAR29.
Otro estudio encontró una correlación entre la extensión de la enfermedad en la TACAR, la mayor duración de la enfermedad y la disminución de la DLCO, el volumen espiratorio forzado (VEF1) y la capacidad vital forzada (CVF)30. Adicionalmente, la presencia de panal de abeja y áreas de vidrio esmerilado en la TACAR se ha correlacionado con una DLCO reducida.
El compromiso intersticial tiende a ser más frecuente en las bases pulmonares, y se define en mayor medida cuando el paciente presenta síntomas respiratorios de forma persistente31. Los patrones radiológicos más comunes de EPI en el LES incluyen la NINE, la NO y la NIL. La NIL se caracteriza por infiltración difusa del intersticio por linfocitos policlonales y suele acompañarse de alveolitis linfocítica.
Con frecuencia se observa atenuación en vidrio esmerilado, nódulos centrolobulillares y engrosamiento septal. Asimismo, se pueden observar quistes pulmonares de tamaño variable32,33. La NO rara vez se ha informado en el LES. Por lo general sigue a un daño pulmonar local, ya sea por infecciones, medicamentos, irradiación o sustancias químicas. Se ha descrito como la manifestación inicial del LES y puede ocurrir con independencia de la actividad de la enfermedad34,35. En la figura 2 se presenta un diagrama con las características de clasificación de las neumonías intersticiales idiopáticas.

Clasificación de las neumonías intersticiales idiopáticas.
BAL:broncoscopia con lavado broncoalveolar; LI:lóbulos inferiores; LS:lóbulos superiores; NIL:neumonía intersticial linfoide; NINE:neumonía intersticial no específica; NIU:neumonía intersticial usual; NO:neumonía en organización.
Fuente: Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733-48.
El lavado broncoalveolar (LBA) se considera un método eficaz y relativamente seguro para obtener el recuento de células inflamatorias y muestras de secreción en pacientes con enfermedad pulmonar. Se ha descrito que incluso en pacientes asintomáticos se presenta una alveolitis subclínica36. Un estudio analizó el fluido del LBA en pacientes no tratados y con diagnóstico reciente de LES. Los porcentajes medios de linfocitos, neutrófilos y macrófagos fueron 23,6, 6,2 y 70,5%, respectivamente. Los pacientes con síntomas pulmonares tuvieron un predominio de linfocitos, mientras que los que presentaban cambios radiográficos consistentes con EPI tuvieron predominio de linfocitos y neutrófilos37. La fibrobroncoscopia flexible con LBA puede ser útil para descartar infecciones, hemorragia y malignidad, pero no parece ser una herramienta útil para el diagnóstico de enfermedad pulmonar intersticial.
DiagnósticoEl diagnóstico de la EPI asociada con LES es fundamentalmente clínico, sustentado por la presencia de manifestaciones extrapulmonares y serológicas en combinación con una TACAR que confirme los hallazgos relacionados de enfermedad intersticial con un patrón radiológico específico. Se deben excluir otras causas de compromiso intersticial como infecciones, toxicidad por medicamentos y falla cardiaca aguda. En raras ocasiones, se requiere una biopsia pulmonar si el diagnóstico no es claro y los resultados de la biopsia transbronquial no son concluyentes38.
TratamientoAl igual que con el tratamiento de otras enfermedades pulmonares intersticiales, se recomienda enfáticamente abandonar el hábito del tabaquismo. La atención de apoyo con oxígeno suplementario está indicada en pacientes con hipoxemia en reposo o inducida por el ejercicio. Las vacunas contra la influenza, el neumococo y la COVID-19 se deben administrar según las pautas de recomendación actual39. No se conoce la terapia óptima para la EPI asociada con LES debido a la escasez de casos informados y la ausencia de ensayos clínicos controlados. Para definir si el paciente amerita tratamiento inmunosupresor se debe evaluar la severidad y la tasa de progresión junto con las indicaciones de tratamiento de las manifestaciones extrapulmonares del LES.
Por lo general, se reserva el tratamiento para pacientes que tengan un patrón radiológico no fibrosante con evidencia de progresión demostrada por una caída de la CVF o DLCO por lo menos del 10%. Con frecuencia, los pacientes que presentan un fenotipo fibrosante progresivo, como es el caso de la NIU o la NINE fibrótica, tienen menos probabilidades de beneficiarse de la terapia inmunosupresora, aunque pueden alcanzar respuestas terapéuticas con la terapia antifibrótica si los inmunosupresores no logran retrasar la progresión de la enfermedad40. Para los patrones radiológicos comunes se indica inicialmente glucocorticoides sistémicos como la prednisona a dosis de 0,5 a 1mg/kg por día.
En un estudio observacional de 14 pacientes con EPI asociada con LES se describió que tres pacientes mostraron una mejoría significativa con dosis altas de glucocorticoides orales (60mg de prednisolona al día durante un mínimo de 4 semanas)41. La elección de un agente ahorrador de glucocorticoides se fundamenta en el grado de insuficiencia respiratoria y las comorbilidades del paciente. En los casos de EPI leve a moderada el micofenolato o la azatioprina son una opción razonable, aunque su evidencia sigue siendo limitada42. En el caso de los pacientes que presentan un brote de la enfermedad o una EPI progresiva y grave (hipoxemia marcada y deterioro importante en las pruebas de función pulmonar), el tratamiento se inicia con dosis altas de glucocorticoides (metilprednisolona intravenosa 1g al día durante 3 días) y ciclofosfamida (generalmente por vía intravenosa) o rituximab con transición a azatioprina o micofenolato después de 6 a 12 meses de tratamiento43,44.
No se ha establecido un tratamiento específico para la enfermedad pulmonar fibrótica en pacientes con LES. Sin embargo, un estudio clínico de nintedanib, un inhibidor de la tirosina quinasa, que incluyó a pacientes con varias EPI fibrosantes progresivas, entre ellas la EPI asociada con enfermedades autoinmunes, mostró una reducción en la tasa de disminución de la CVF45,46. La dosis de nintedanib es de 150mg, 2 veces al día, con aproximadamente 12 h de diferencia. Se deben tener pruebas de función hepática antes de su inicio dado que está contraindicado en pacientes con insuficiencia hepática moderada o severa (Child Pugh B o C). Es importante destacar que el nintedanib puede causar daño fetal. Por tanto, se debe tener una prueba de embarazo negativa antes de iniciar el tratamiento en mujeres con potencial reproductivo, y se debe usar un método anticonceptivo altamente efectivo durante la terapia47.
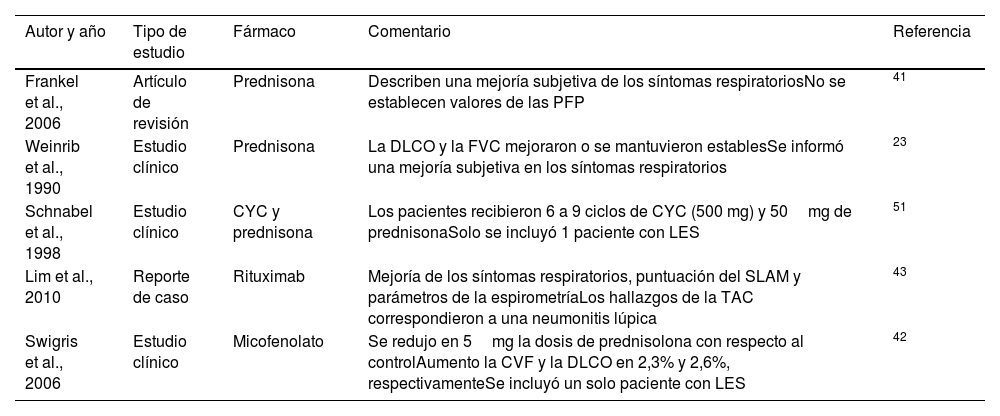
El trasplante de pulmón puede ser la última opción de tratamiento para pacientes seleccionados con EPI fibrosante avanzada48. En las figuras 3 y 4 se presentan dos diagramas que exponen el orden jerárquico del tratamiento de la EPI asociada con LES. De la misma manera, en la tabla 1 se relacionan los artículos más relevantes que sustentan las recomendaciones de las líneas de tratamiento incluidas en los mencionados diagramas.


Artículos más representativos para establecer las líneas de tratamiento en orden jerárquico
| Autor y año | Tipo de estudio | Fármaco | Comentario | Referencia |
|---|---|---|---|---|
| Frankel et al., 2006 | Artículo de revisión | Prednisona | Describen una mejoría subjetiva de los síntomas respiratoriosNo se establecen valores de las PFP | 41 |
| Weinrib et al., 1990 | Estudio clínico | Prednisona | La DLCO y la FVC mejoraron o se mantuvieron establesSe informó una mejoría subjetiva en los síntomas respiratorios | 23 |
| Schnabel et al., 1998 | Estudio clínico | CYC y prednisona | Los pacientes recibieron 6 a 9 ciclos de CYC (500 mg) y 50mg de prednisonaSolo se incluyó 1 paciente con LES | 51 |
| Lim et al., 2010 | Reporte de caso | Rituximab | Mejoría de los síntomas respiratorios, puntuación del SLAM y parámetros de la espirometríaLos hallazgos de la TAC correspondieron a una neumonitis lúpica | 43 |
| Swigris et al., 2006 | Estudio clínico | Micofenolato | Se redujo en 5mg la dosis de prednisolona con respecto al controlAumento la CVF y la DLCO en 2,3% y 2,6%, respectivamenteSe incluyó un solo paciente con LES | 42 |
CVF:capacidad vital forzada; CYC:ciclofosfamida; DLCO:prueba de difusión de monóxido de carbono; PFP:pruebas de función pulmonar; SLAM:Systemic Lupus Activity Measure.
El seguimiento se debe realizar a intervalos de 3 a 6 meses con PFP y evaluar algún cambio en los síntomas o en la exploración física. Se puede disminuir la frecuencia de las PFP cada 2 años en pacientes que tengan una DLCO normal, no presenten disnea y las PFP se mantengan sin cambios durante más de 3 años. Se puede considerar hacer una nueva TACAR si el paciente desarrolla nuevos síntomas respiratorios o si se produce una disminución en los volúmenes pulmonares o de la DLCO49.
PronósticoLa mayoría de los pacientes en las series descritas que han sido tratados con glucocorticoides sistémicos han logrado una mejoría en la DLCO y la CVF o se han mantenido estables con mejoría de la disnea, el dolor pleurítico y la tos crónica. A pesar de esto, el curso clínico de la EPI en el LES es variable, aunque suele tener una progresión lenta con una tendencia a mejorar o estabilizarse en el tiempo50. Sin embargo, el pronóstico también dependerá del tipo de patrón radiológico establecido, dado que un patrón de NIU conllevará probablemente una mayor progresión y un curso posiblemente irreversible.
ConclusionesEn la EPI, a pesar de ser un hallazgo infrecuente en el LES, los casos moderados a severos o los que presentan un fenotipo fibrosante progresivo representan un gran desafío terapéutico, debido a los pocos casos reportados en la literatura, por lo que muchas estrategias de tratamiento han sido extrapoladas de la EPI asociada a la esclerosis sistémica. Los glucocorticoides siguen siendo el pilar fundamental del tratamiento y la consideración del inicio conjunto de un ahorrador de glucocorticoides dependerá de las comorbilidades del paciente y la severidad de la enfermedad. En los casos de brote o enfermedad progresiva severa se deberá emplear ciclofosfamida o rituximab con posterior transición a la terapia de mantenimiento. La terapia antifibrótica tiene indicación en cualquier tipo de patrón radiológico en el contexto de enfermedades autoinmunes, aunque se desconoce el momento óptimo de inicio y su duración.
Es probable que la terapia antifibrótica sea más eficiente en los pacientes que tienen una EPI con fenotipo fibrosante progresivo para evitar la caída de la CVF. Sin embargo, la mayoría de los estudios no han evaluado la mortalidad como desenlace primario. Si bien es cierto que la mayoría de los pacientes con EPI asociado con LES tienen un mejor pronóstico con respecto a los casos de fibrosis pulmonar idiopática, los pacientes que presentan un fenotipo fibrosante progresivo y falla terapéutica requieren derivación a un centro especializado para considerar el trasplante pulmonar.
FinanciaciónEste artículo no ha recibido financiación específica del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
