






La esclerosis lateral amiotrófica (ELA) es la enfermedad degenerativa de las motoneuronas más frecuente. Aunque un pequeño porcentaje de los casos de ELA tienen un origen familiar y son secundarios a mutaciones en genes concretos, a la gran mayoría de ellos se les presupone un origen multifactorial, sin que su patogenia haya sido completamente aclarada. No obstante, en los últimos años varios estudios han aumentado el conocimiento sobre la patogenia de la enfermedad, planteando la cuestión de si se trata de una proteinopatía, una ribonucleinopatía, una axonopatía o una enfermedad del microambiente neuronal.
DesarrolloEn el presente artículo revisamos los trabajos publicados tanto en pacientes como en modelos animales de ELA y discutimos la implicación de los principales procesos celulares que parecen contribuir a su patogenia (procesamiento génico, metabolismo de proteínas, estrés oxidativo, transporte axonal y relación con el microambiente neuronal).
ConclusionesAunque la patogenia de la ELA dista de estar aclarada, los estudios recientes apuntan a la idea de que hay unos desencadenantes iniciales que varían de unos sujetos a otros, y unas vías finales de degeneración de las motoneuronas que están implicadas en la mayor parte de los casos de enfermedad.
Amyotrophic lateral sclerosis (ALS) is the most common neurodegenerative disease affecting motor neurons. Although a small proportion of ALS cases are familial in origin and linked to mutations in specific genes, most cases are sporadic and have a multifactorial aetiology. Some recent studies have increased our knowledge of ALS pathogenesis and raised the question of whether this disorder is a proteinopathy, a ribonucleopathy, an axonopathy, or a disease related to the neuronal microenvironment.
DevelopmentThis article presents a review of ALS pathogenesis. To this end, we have reviewed published articles describing either ALS patients or ALS animal models and we discuss how the main cellular pathways (gene processing, protein metabolism, oxidative stress, axonal transport, relationship with neuronal microenvironment) may be involved in motor neurons degeneration.
ConclusionsALS pathogenesis has not been fully elucidated. Recent studies suggest that although initial triggers may differ among patients, the final motor neurons degeneration mechanisms are similar in most patients once the disease is fully established.
La esclerosis lateral amiotrófica (ELA) es la enfermedad degenerativa de las motoneuronas (MN) más frecuente, con una incidencia que oscila entre 1 y 3 casos por cada 100.000 habitantes y año1,2. Aunque hay casos descritos a partir de la segunda y tercera décadas de la vida, la incidencia máxima de enfermedad se sitúa entre los 60 y 70 años3. La ELA se caracteriza por una afectación conjunta de la MN superior y de la MN inferior que conducirá a la aparición de la sintomatología clínica, con debilidad muscular, atrofia, fasciculaciones y espasticidad. Dentro de los casos de ELA se distinguen dos grandes grupos: un primer grupo que representa entre un 5 y un 10% de los casos en el que los pacientes afectos muestran agregación familiar, habitualmente con un patrón de transmisión mendialiana autosómica dominante (ELA familiar [ELAf]) y por otro lado, aquellas otras formas en las que no se encuentra una clara historia familiar y a las que se le presuponen un origen esporádico (ELA esporádica [ELAe])3. Las primeras son secundarias a mutaciones en genes directamente relacionados con la degeneración de las MN, mientras que en las formas esporádicas se presupone un origen multifactorial. En el presente trabajo pretendemos revisar las diferentes rutas celulares disfuncionantes en la ELA y cómo su alteración predispone a la degeneración de las MN.
DesarrolloLa ELA representa una alteración específica de tejido y de tipo celular, siendo las MN las principales células afectadas (fig. 1). Aunque los factores que determinan la afectación preferente de este tipo neuronal no son bien conocidos4, se asumen como factores para su especial vulnerabilidad: a) su gran tamaño, con un citoesqueleto muy desarrollado, lo que implica una gran actividad metabólica para mantener las funciones celulares, b) unos requerimientos mitocondriales muy elevados, c) su elevada sensibilidad a los agentes excitotóxicos, así como a las alteraciones en la regulación del calcio intracelular y d) una capacidad reducida de respuesta de «choque térmico» y de procesos chaperonodependientes, así como de la función del sistema ubiquitina-proteasoma4. Aunque los mecanismos patogénicos subyacentes en la ELA son múltiples y no se han aclarado completamente; se han caracterizado y relacionado con la aparición de la enfermedad una serie de factores genéticos y alteraciones en las principales rutas celulares que serán comentados a continuación (fig. 2).


Alteraciones patogénicas descritas en la ELA. En la ELA se han descrito alteraciones a múltiples niveles de las funciones celulares, las cuales se ven recogidas en la presente figura. Se han identificado alteraciones a nivel del procesamiento de los ARN consecuencia de las cuales aparecen ARN aberrantes y/o ARN tóxicos. Niveles elevados de estrés oxidativo así como dificultades en la eliminación de los radicales libres también se han relacionado con esta enfermedad. Por otro lado aparecen alteraciones en el metabolismo de las proteínas con inhibición/malfunción del SUP y una hiperactivación de la autofagia. Desórdenes en proteínas implicadas en el transporte axonal también se han descrito como causa de ELA. Por último se han reportado alteraciones a nivel de las células gliales que repercuten en las MN conduciendo a la degeneración de las mismas.
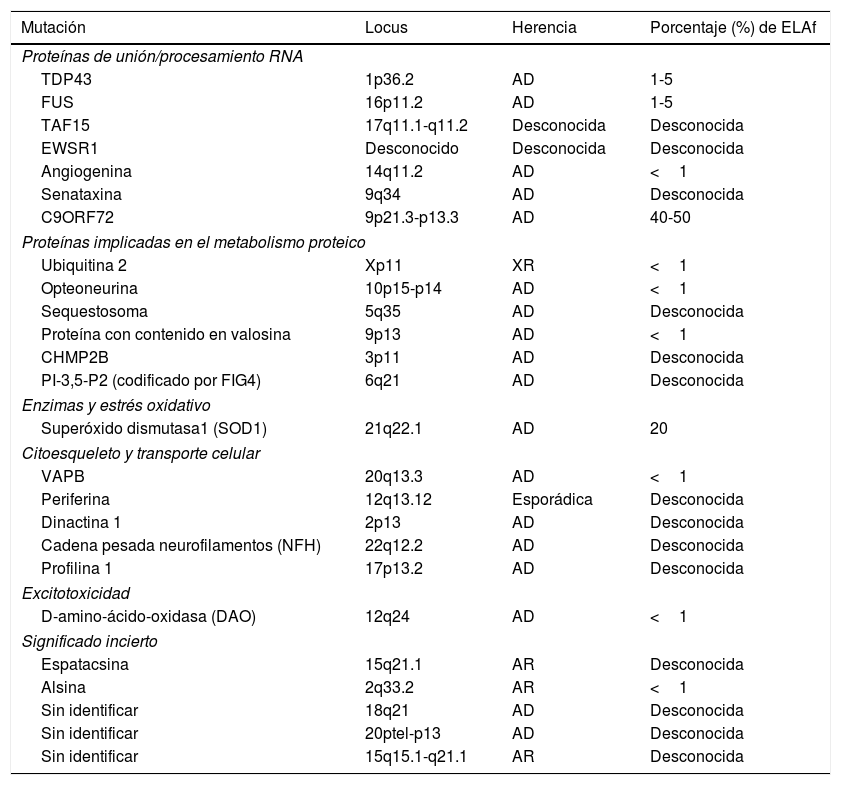
Las formas de ELA familiar (ELAf) suponen un 5-10% de los casos, presentando habitualmente una herencia autosómica dominante, siendo las más frecuentes las secundarias a mutaciones en el gen de la superóxido dismutasa 1 (SOD1) y en el cromosoma 9. En la tabla 1 se recogen los diferentes genes identificados en pacientes con ELAf hasta el momento actual. El estudio de los casos familiares es muy importante ya que permite el planteamiento de nuevas hipótesis etiopatogénicas extrapolables a los casos de ELAe4.
Genes identificados en formas de ELA familiar
| Mutación | Locus | Herencia | Porcentaje (%) de ELAf |
|---|---|---|---|
| Proteínas de unión/procesamiento RNA | |||
| TDP43 | 1p36.2 | AD | 1-5 |
| FUS | 16p11.2 | AD | 1-5 |
| TAF15 | 17q11.1-q11.2 | Desconocida | Desconocida |
| EWSR1 | Desconocido | Desconocida | Desconocida |
| Angiogenina | 14q11.2 | AD | <1 |
| Senataxina | 9q34 | AD | Desconocida |
| C9ORF72 | 9p21.3-p13.3 | AD | 40-50 |
| Proteínas implicadas en el metabolismo proteico | |||
| Ubiquitina 2 | Xp11 | XR | <1 |
| Opteoneurina | 10p15-p14 | AD | <1 |
| Sequestosoma | 5q35 | AD | Desconocida |
| Proteína con contenido en valosina | 9p13 | AD | <1 |
| CHMP2B | 3p11 | AD | Desconocida |
| PI-3,5-P2 (codificado por FIG4) | 6q21 | AD | Desconocida |
| Enzimas y estrés oxidativo | |||
| Superóxido dismutasa1 (SOD1) | 21q22.1 | AD | 20 |
| Citoesqueleto y transporte celular | |||
| VAPB | 20q13.3 | AD | <1 |
| Periferina | 12q13.12 | Esporádica | Desconocida |
| Dinactina 1 | 2p13 | AD | Desconocida |
| Cadena pesada neurofilamentos (NFH) | 22q12.2 | AD | Desconocida |
| Profilina 1 | 17p13.2 | AD | Desconocida |
| Excitotoxicidad | |||
| D-amino-ácido-oxidasa (DAO) | 12q24 | AD | <1 |
| Significado incierto | |||
| Espatacsina | 15q21.1 | AR | Desconocida |
| Alsina | 2q33.2 | AR | <1 |
| Sin identificar | 18q21 | AD | Desconocida |
| Sin identificar | 20ptel-p13 | AD | Desconocida |
| Sin identificar | 15q15.1-q21.1 | AR | Desconocida |
Entre las mutaciones génicas se incluyen la del factor de crecimiento del endotelio vascular y la proteína de la hemocromatosis hereditaria. También se han descrito variaciones en el número de copias en los genes 1 y 2 que codifican el factor de supervivencia de las MN (SMN1 y SMN2)5. No obstante, la correlación con la ELA no ha podido ser reproducida por completo en estudios posteriores realizados en otros grupos de población. Por otro lado, el estudio de genes candidatos en pacientes con formas esporádicas ha identificado distintos polimorfismos asociados a un mayor riesgo de enfermedad. Hasta la fecha se han publicado varios estudios de asociación del genoma completo (en inglés Genome-wide association study) en la ELA. En ellos se han identificado una serie de loci relacionados con una mayor susceptibilidad para el desarrollo de la enfermedad, como son la carbohidrato kinasa FGGY (FGGY), la dipeptidil-peptidasa 6 (DPP6) o el receptor tipo 2 del inositol trifosfato6–8. Sin embargo, la mayoría de estos hallazgos no han conseguido reproducirse posteriormente en grandes cohortes de pacientes. La relación patogénica de otros genes, como el homólogo A de la proteína de unión a las vesículas sinápticas unc-13 (UNC13B), parece más robusta, aunque todavía se encuentra pendiente de ser reproducido en otros estudios9. En 2010 se identificó que una expansión intrónica en el gen C9ORF72, en una importante proporción de casos de ELA, tanto esporádicos como familiares, que hasta entonces se clasificaba como ELA asociada al cromosoma 910,11. Característicamente, esta expansión se ha descrito también en pacientes con demencia frontotemporal (DFT) y en pacientes con el complejo ELA-DFT12,13. Dada la gran relevancia que ha cobrado este hallazgo, será comentado en profundidad más adelante.
Hasta el momento, los estudios genéticos se han centrado principalmente en poblaciones de origen europeo. En el futuro, la realización de estudios genéticos en pacientes de otras etnias así como el análisis variantes raras, esto es, polimorfimsos con frecuencia alélica <5%, posibilitará el descubrimiento de nuevos loci patogénicos. Recientemente, el análisis del exoma ha mostrado mutaciones de novo en algunos genes implicados principalmente en formas de ELAf, como las secundarias a mutaciones de SOD y «fused in sarcoma» (FUS); si bien es cierto que no todas ellas son necesariamente patogénicas14–16. El estudio del exoma es un campo emergente, con una gran proyección, lo que seguramente aportará nueva información en los próximos años.
Alteraciones transcripcionales y en el procesamiento de los ARNUn gran número de proteínas que se ha relacionado con la ELA se encuentran directa o indirectamente implicadas en el procesamiento de los ácidos nucleicos. Los primeros hallazgos surgieron tras la identificación de mutaciones en el gen SMN1 como causa de la atrofia muscular espinal17. La SMN es una proteína vital para la supervivencia de las MN. Esta proteína está implicada en el «splicing» (proceso de corte y empalme) de los pre-ARNm18 y en el transporte axonal de los ARN mensajeros para su posterior traducción en el terminal presináptico, particularmente en la placa motora18,19.
Posteriormente, la identificación de mutaciones en TDP43 y FUS confirmó que un metabolismo aberrante del los ARN podía contribuir a la patogenia de la ELA.
Proteína 43 de unión a DNA-TAR (TDP43)Las mutaciones en el gen de TARDBP que codifica para la proteína TDP43 («TAR-DNA binding protein 43») son una causa rara de ELAf, que presenta una herencia autosómica dominante20. La investigación de TDP43 como gen candidato surgió tras identificar esta proteína en las inclusiones neuronales de la mayoría de los pacientes con ELA, tanto esporádica como familiar21. Llamativamente, estas inclusiones no aparecen en los casos secundarios a mutaciones en SOD y en FUS21. TDP43 es una proteína de la familia de las ribonucleoproteínas que se encuentra predominantemente en el núcleo y que interacciona con las moléculas de ADN y ARN, interviniendo en la transcripción, «splicing» y posterior transporte de los ARN22. La mayoría de las mutaciones en el gen TARDBP se encuentran en el exón 6, tratándose de mutaciones sin sentido23. La proteína contiene dos dominios «prion like» de interacción, que se unen a las regiones intrónicas ricas en GU de las moléculas de ARN, pudiendo interaccionar con más de 6.000 ARN mensajeros distintos22. En situaciones de estrés TDP43 se encuentra predominantemente en el citoplasma, formando parte de agregados citoplasmáticos conocidos como «gránulos de estrés»24. En un intento de priorizar la traducción de aquellas proteínas esenciales para la supervivencia celular, estos agregados secuestran los ARN no implicados en la respuesta de estrés neuronal, bloqueando su traducción, hasta que el insulto estresante cesa. Cuando esto ocurra, los agregados se disociarán por la acción de peptidasas y chaperonas, y TDP43 retornará al compartimento nuclear25. Los mecanismos por los cuales las mutaciones en TDP43 conducen al desarrollo de la ELA no se han aclarado por completo. Las mutaciones asociadas al desarrollo de la enfermedad aumentan la translocación de la proteína del núcleo al citoplasma y potencian su tendencia a la agregación. Por lo tanto, la proteína anormal no podrá cumplir sus funciones habituales en la transcripción y en el procesamiento de ARN, creando además un ambiente proagregante en el citoplasma que tenderá a secuestrar otras moléculas necesarias para la homeostasis celular22,26,27.
Familia de proteínas de la familia FET: la proteína FUSOtros genes relacionados con el procesamiento génico y también implicados en la patogenia de la ELA son los que codifican la familia de proteínas FET. La familia de FET está constituida por la proteína TAF15 («TATA binding protein associated factor 15»), «Ewing sarcoma breakpoint region 1»(EWSR1) y la proteína FUS28.
FUS es una proteína nuclear de unión a ADN y ARN implicada en la regulación transcripcional, el procesamiento de ADN y ARN y en el transporte de los ARN mensajeros. Las mutaciones en FUS suponen un 4% de los casos de ELAf y menos de un 1% de los de ELAe21. La mayoría de las mutaciones en FUS se encuentran en los exones 13 y 15, que codifican una secuencia de localización nuclear. Estas mutaciones van a provocar una traslocación anómala al citoplasma, donde tenderá a reclutarse en gránulos de estrés29,30. Al igual que sucede con TDP43, FUS interacciona con muchos genes, posiblemente más de 5.500, a través de un dominio de unión «prion like», por lo que potencialmente parece influir en un gran número de procesos celulares31. Aunque los mecanismos por los cuales FUS resulta citotóxica no han sido aclarados; es probable que coexistan fenómenos de pérdida y ganancia de función, al igual que ocurre con TDP43.
Mutaciones en el gen C9ORF72El gen C9ORF72 contiene una secuencia hexanucleótida GGGGCC que se localiza entre dos lugares de inicio de la transcripción. Una expansión anormal de esta secuencia se ha encontrado hasta en un 40% de las familias con ELA y en un 7% de los pacientes con ELAe32. Como se ha comentado anteriormente, esta expansión ha sido también documentada en pacientes con DFT con y sin ELA asociada10,32. Normalmente los individuos sanos presentan de 2 a 5 repeticiones y nunca más de 30. La función de la proteína C9ORF72 es desconocida, se sabe que esta proteína se encuentra estructuralmente relacionada con la familia de proteínas con dominio «differentially expressed in normal and neoplastic cells»33. Funcionalmente, el dominio «differentially expressed in normal and neoplastic cells» está implicado en la regulación de una serie de GTPasas, enzimas que utilizan y degradan GTP34. La penetrancia de las mutaciones es del 50% a los 60 años, situándose cercana al 100% pasados los 80 años32. El mecanismo patogénico por el cual el exceso de repeticiones se asocia con el desarrollo de la enfermedad aún no ha sido aclarado; existiendo numerosas cuestiones como si el carácter homocigoto o un mayor número de repeticiones se asocian a fenotipos más agresivos por estudiar. Algunos estudios en pacientes con la expansión han demostrado cómo los niveles de ARN mensajeros de C9ORF72 estaban reducidos hasta en un 50%, pudiendo inferir que probablemente el alelo mutado no es capaz de generar un ARN maduro, lo que estaría en relación con un proceso de pérdida de función35. Por otro lado, estudios de hibridación in situ en pacientes con ELA portadores de la expansión demostraron la presencia de agregados de ARN nuclear en la corteza y la médula espinal sugiriendo asimismo un mecanismo de ganancia tóxica de función10.
Otras proteínas implicadas en el procesamiento génicoAunque de menor importancia, en los pacientes con ELA se han descrito también algunas alteraciones en otras proteínas implicadas en el procesamiento génico como la angiogenina y la senataxina36.
Alteraciones en el metabolismo proteicoLa síntesis de proteínasEl nucleolo y el retículo endoplásmico (RE), cuyas funciones son la síntesis de ARN ribosomales y proteínas, respectivamente, así como los sistemas ubiquitina-proteasoma (SUP) y lisosomal- autofágico, implicados en el aclaramiento/eliminación de proteínas, son los principales elementos implicados en el metabolismo proteico de la célula. La mayoría de las enfermedades neurodegenerativas se caracterizan por la presencia de agregados celulares de proteínas aberrantes o mal plegadas. En el caso concreto de la ELA, se postula que dichos agregados y sus precursores oligoméricos alteran el normal funcionamiento celular, induciendo un grado de estrés oxidativo que resulta perjudicial para la célula, ya que interfiere con sus funciones básicas y conduce finalmente a la muerte neuronal37.
La síntesis de proteínas, así como su plegamiento y «control de calidad», constituyen algunas de las funciones importantes del RE. En determinadas condiciones de estrés celular se induce una respuesta de «estrés del RE» que se caracteriza por la presencia de cromatólisis y por el acúmulo de proteínas mal plegadas y aberrantes en inclusiones citoplasmáticas. El término cromatólisis se refiere a la fragmentación y posterior disolución citoplasmática de las cisternas de RE. Varios trabajos en la literatura han descrito la existencia de cromatólisis y de agregados proteicos, tanto en pacientes como en modelos experimentales de ELA38–41 (fig. 3). La disgregación del RE se traduce en una alteración de su capacidad de síntesis proteica que, en numerosas ocasiones, acabará induciendo la activación de los mecanismos de muerte celular programada39. Los ribosomas constituyen una parte fundamental del RE granular, por lo que su biogénesis es imprescindible para el mantenimiento estructural y funcional de esta organela. La síntesis de ribosomas es un proceso estrictamente regulado mediado por el nucléolo42–44. Es por ello por lo que el nucleolo juega un papel central en la coordinación de las respuestas de estrés del RE45–48. Las alteraciones en la función nucleolar se han relacionado con algunas enfermedades neurodegenerativas49–53. Algunos autores han mostrado en un modelo transgénico de ELA un aumento en el diámetro nucleolar probablemente en relación con un intento celular de aumentar la síntesis de genes ribosomales en situaciones de estrés54.
 y de un modelo de ratón transgénico de ELA (modelo SOD1G93A) teñidas con ubiquitina. Puede observarse cómo en el ratón sano no se observan inclusiones citoplasmáticas, mientras que el ratón transgénico (B) presenta múltiples agregados poliubiquitilados secundarios al colapso proteasomal.")
Se muestran 2 MN de un ratón control sano (A) y de un modelo de ratón transgénico de ELA (modelo SOD1G93A) teñidas con ubiquitina. Puede observarse cómo en el ratón sano no se observan inclusiones citoplasmáticas, mientras que el ratón transgénico (B) presenta múltiples agregados poliubiquitilados secundarios al colapso proteasomal.
En los últimos años se han descrito nuevas formas de ELAf secundarias a mutaciones en genes que codifican proteínas implicadas directamente en la proteostasis. El SUP y el sistema autofagia-lisosomal son las dos vías proteolíticas principales en la célula. Debido al elevado solapamiento funcional que existe entre ambas vías de degradación proteica, resulta difícil determinar cuál de las dos es la principal responsable de la disfunción celular. Algunos trabajos experimentales han intentado «disecar» ambas vías en modelos murinos. Estas investigaciones documentaron cómo la inhibición del proteasoma se correlacionó con la pérdida neuronal y la aparición de agregados celulares ricos en TDP43, OPTN y FUS, mientras que dichos efectos celulares no se observaron al inhibir la autofagia55. Estos resultados sugieren que la homeostasis de las motoneuronas en la ELA podría verse más comprometida en los casos de disfunción del SUP que en desórdenes de la autofagia. Sin embargo, esta extrapolación debe ser tomada con cautela, ya que serán necesarias más investigaciones para clarificar y confirmar estos hallazgos. En cualquier caso, actualmente existen evidencias sólidas de que la disfunción de la proteostasis secundaria a un malfuncionamiento del SUP y la autofagia contribuye a la pérdida de la homeostasis neuronal. A continuación se exponen alguno de los genes implicados en las vías de degradación proteica que se han identificado en los pacientes con ELA.
La ubiquilina-2La UBQLN2 es una proteína similar a la ubiquitina que interviene en la proteostasis promoviendo la proteólisis proteasomal56. Años atrás, mutaciones en esta proteína se demostraron en algunos casos de enfermedad de Alzheimer57. Recientemente se ha descubierto que las mutaciones en la UBQLN2 juegan un papel patogénico en algunos casos de ELA y de ELA-DFT esporádicos, demostrando inclusiones de UBQLN2 en las MNs del asta anterior de pacientes con ELA. Estas inclusiones se han documentado también en pacientes con ELA sin que presentaran propiamente alteraciones en el gen de la UBQLN2, lo que sugiere que la UBQLN2 podría jugar un papel importante en los mecanismos finales de degeneración de las MN58.
Proteína con contenido en valosinaLos estudios de secuenciación del exoma han identificado en un conjunto de pacientes afectos de ELAf mutaciones sin sentido en el gen que codifica la proteína con contenido en valosina (valosin containing protein [VCP])59. La VCP es una chaperona implicada en múltiples procesos biológicos, incluyendo la proteostasis60. En lo referente a esta, la VCP está implicada en la degradación a nivel proteasomal y autofágico61.
CHMP2B, optoneurina y fofatasa PI-3,5-P2Mutaciones en los genes proteína cargada 2b del cuerpo multivesicular (del inglés charged multivesicular body protein 2b [CHMP2B], optoneurina y fosfatidilinositol 3,5-bifosfato 5 fosfatasa (PI-3,5-P2) se han implicado en casos infrecuentes de ELAf62–64. La CHMP2B es una proteína implicada en el aclaramiento autofágico65. Por su parte, la optoneurina regula diversos procesos celulares, entre los que se incluyen el transporte a través de membranas y la secreción de proteínas66. Por último, se han publicado en un pequeño grupo de pacientes con ELA mutaciones en el «factor induced gene 4» (FIG4), que codifica la enzima fosfatidilinositol 3,5-bifosfato 5 fosfatasa. Esta enzima se localiza en la membrana de los endosomas, donde media en la función lisosomal67.
El papel de la SOD 1 y el estrés oxidativoLa enzima SOD1 es una enzima citosólica de 150 aminoácidos con un átomo de cobre y uno de cinc que cataliza la conversión de superóxido en oxígeno y peróxido de hidrógeno lo que le confieren un papel clave en la defensa antioxidante en las células con metabolismo aeróbico, como es el caso de las MN. De hecho, las mutaciones en la enzima SOD1 suponen un 20% de los casos de ELAf. Se han documentado más de 150 mutaciones distintas, la mayoría de ellas sin sentido, presentando casi en su totalidad una herencia autosómica dominante23. Las mutaciones de SOD1 provocan importantes desórdenes en su plegamiento, lo que provoca que sea procesada para su degradación por el SUP. Sin embargo, una gran proporción de la proteína mal plegada no va a poder ser eliminada por SUP, lo cual implicará una interferencia con la proteostasis celular68,69 produciendo una activación autofágica secundaria. En esta línea, se ha demostrado un aumento de autofagosomas, tanto en modelos murinos como en pacientes con ELA70,71. La acumulación progresiva de la proteína mutada mal plegada, conjuntamente con la acción de otros factores estresantes, como el envejecimiento, inducirá la aparición de una respuesta de estrés celular72. Las alteraciones en el plegamiento de SOD1 no son exclusivas de las formas mutantes. Se ha comprobado que en condiciones de estrés oxidativo, la proteína SOD1 nativa tiende al plegamiento aberrante y a la agregación, de manera similar a lo que ocurre con la proteína mutada73,74. Estos hallazgos sugieren que la proteína SOD1 podría tener también un papel en las formas de enfermedad esporádica, contribuyendo a su patogenia una vez que el estrés celular hubiese sido instaurado por otros mecanismos.
Los estados de estrés oxidativo aparecen cuando existe un desequilibrio entre la generación y eliminación de los radicales libres, o cuando existe una incapacidad en la célula para reparar o eliminar los daños secundarios a dicho estrés. Diversas evidencias sugieren su implicación en la patogenia ELA. Así pues, el análisis de muestras de sangre, orina y líquido cefalorraquídeo de pacientes con ELAe ha mostrado una elevación en marcadores de daño por radicales libres75–77. Por otro lado, los estudios necrópsicos también han confirmado la existencia de alteraciones proteicas, lipídicas y del ADN secundarias a radicales de oxígeno tanto en pacientes con ELAe como en pacientes con ELAf secundaria a mutaciones en SOD178–80. Por su parte, los estudios en modelos murinos de SOD1 mutante han documentado cómo en etapas presintomáticas aparecen niveles elevados de oxidación de distintos ARN mensajeros y cómo esta oxidación se correlaciona con una menor expresión posterior de la proteína. Así, la hipótesis más extendida actualmente postula que, además del daño que provoca en sí mismo, el estrés oxidativo va a potenciar otros mecanismos patogénicos que contribuyen a la alteración neuronal, entre los que se incluyen la excitotoxicidad, la agregración de proteínas, la respuesta de estrés de RE y la disfunción mitocondrial así como a las relaciones de las MN con el microambiente neuronal.
Alteraciones en el transporte axonalLas MN son células con axones de gran longitud por lo que el transporte axonal constituye un aspecto clave en su biología. Dicho transporte, dependiente de ATP, es vital para el abastecimiento de los componentes celulares necesarios (ARN, proteínas y diversas organelas) al terminal presináptico. La maquinaria principal para el transporte axonal está constituida por los microtúbulos y las proteínas motoras, kinesinas y dineínas, asociadas a los microtúbulos72,81. Diversos estudios en ratones transgénicos y pacientes con ELA han demostrado que la desnervación y la retracción axonal ocurren en estadios de enfermedad muy tempranos, antes incluso de que la pérdida de cuerpos neuronales se haga evidente82,83. En el modelo murino de SOD1 se ha observado cómo en etapas iniciales se alteran tanto el transporte anterógrado como el retrógrado, y cómo esta alteración es cargo-dependiente, encontrándose por ejemplo especialmente comprometido el transporte mitocondrial anterógrado83–85. Clásicamente, se ha considerado que las alteraciones en el transporte axonal son secundarias a desórdenes múltiples: las alteraciones mitocondriales y consecuentemente una producción insuficiente de ATP, disrupción en las funciones de las kinesinas, acúmulo de agregados proteicos etc.86–88. Sin embargo, en los últimos años también se han identificado alteraciones en genes que codifican proteínas directamente implicadas en el transporte axonal. En algunos pacientes con ELA se ha observado una disminución en la expresión de «neurofilament light chain»89. Llamativamente, algunos autores han descrito cómo el ARN mensajero de «neurofilament light chain» tiende a quedar reclutado en los gránulos de estrés de las MN de pacientes con ELA90. Otro de los hallazgos recientes ha sido la descripción de mutaciones en el gen de la PFN1 (profilina 1)91. La PFN1 es esencial para la polimerización de los filamentos de actina, por lo que las mutaciones en este gen provocan una inhibición en el crecimiento axonal, aumentando la tendencia a la retracción axonal y la desnervación. También se han asociado con algunos casos de ELA ciertas mutaciones en los genes que codifican la «neurofilament heavy chain», la periferina, la dinactina y la alsina entre otras92–95. En contraposición, aquellos factores que promueven el crecimiento y la regeneración axonal, como el factor de crecimiento endotelial, han mostrado indicios de neuroprotección96,97.
Por último, en esta línea cabe reseñar que en los últimos años los estudios de GWAS han mostrado relación entre casos de ELAe y algunos polimorfismos en genes que codifican para proteínas axonales como la proteína 3 asociada a kinesina o unas subunidades del complejo acetiltransferasa98,99.
La excitotoxicidadEl glutamato es el principal neurotransmisor excitador en el SNC. Tras la liberación del glutamato y su interacción con el receptor postsináptico, los estímulos excitadores desaparecen con la retirada del neurotransmisor de la hendidura sináptica, gracias a los transportadores recaptadotes de glutamato. Dentro del conjunto de transportadores, el «excitatory amino acid transporter 2» es el más abundante. La excitotoxicidad por hiperactivación de los receptores glutamatérgicos puede ser consecuencia de un incremento en los niveles sinápticos del neurotransmisor o de una mayor sensibilidad al glutamato en el terminal postsináptico100. Los receptores AMPA median gran parte de la neurotransmisión glutamatérgica en el SNC. La permeabilidad al calcio de la familia de receptores AMPA está en gran parte determinada por una subunidad proteica conocida como GluR2. Las MN presentan una especial sensibilidad a la excitotoxicidad secundaria a la activación de los receptores AMPA, ya que presentan una expresión disminuida tanto de la subunidad GluR2 como de proteínas tamponadoras para el ión calcio101,102. Hasta la fecha, varios estudios sugieren que la excitotoxicidad juega un papel patogénico en la ELA. En algunos pacientes con ELA se han detectado niveles elevados de glutamato en LCR, así como una disminución en la expresión de «excitatory amino acid transporter 2» en las áreas del SNC afectadas103,104. Otros estudios han demostrado estados de hiperexcitabilidad motora en estadios presintomáticos de enfermedad, junto con una permeabilidad anormal al calcio de los receptores AMPA105,106. Aunque resulta difícil diferenciar si el daño excitotóxico se trata de un mecanismo patogénico primario o si es una consecuencia del fracaso en la homeostasis neuronal, parece que una vez instaurado el cuadro, las alteraciones en la homeostasis neuronal contribuyen a la progresión del mismo.
La neuroinflamación y el papel de las células glialesAunque la pérdida y la degeneración de las MN es el hallazgo principal en los pacientes con ELA, existe, de manera concomitante, una activación de la respuesta neuroinflamatoria. De tal manera, estudios histológicos de la médula espinal de pacientes con ELA han objetivado la presencia de microglía activada e infiltrados linfocíticos107. Dicha respuesta inflamatoria contribuye a la degeneración y alteración fenotípica de las MN desde las primeras manifestaciones de enfermedad, viéndose incrementada a medida que esta va progresando108,109. Por su parte, los estudios de muestras biológicas en pacientes con ELA han mostrado la presencia de mediadores inflamatorios, con aumento de la interleucina 8 en LCR, así como parámetros sugestivos de activación de la respuesta inmune a nivel de sangre periférica110.
Los astrocitos son células de estirpe glial que se encuentran en íntima asociación con las MN. Algunos trabajos han documentado que estos también pueden inducir degeneración de las MN. Así, se ha comprobado cómo los astrocitos SOD1 mutantes activados secretan una serie de mediadores inflamatorios, entre los que se encuentran la prostaglandina E2, el leucotrieno B4 y el óxido nítrico, que resultan dañinos para las MN111. Estudios posteriores han demostrado también cómo los astrocitos de pacientes con ELAe no portadores de mutaciones en SOD1 presentan un efecto tóxico similar112. Aunque en menor medida, también se han implicado los oligodendrocitos en la patogenia de las enfermedades de la MN. Los oligodendrocitos, además de formar la mielina de las fibras nerviosas del SNC, contribuyen a regular el metabolismo axonal, reduciendo la concentración de ácido láctico neuronal gracias al transportador de lactato MCT1113. Algunos autores han mostrado cómo la pérdida de dicho transportador resulta tóxica para las MN. Asimismo, se ha constatado una disminución en la expresión del transportador tanto en pacientes con ELA como en el modelo murino SOD1114.
ConclusionesEn resumen, cada vez son mayores las evidencias que apoyan que la ELA no es una enfermedad sino un síndrome clínico que se caracteriza por una degeneración de ambas MN y que comparte una sintomatología clínica característica. Dentro de los casos de ELA existe un pequeño porcentaje de casos con herencia familiar y que son secundarios a mutaciones en genes concretos cuya alteración en sí misma, induce la degeneración de las MN. La etiopatogenia de las formas esporádicas no ha sido aclarada del todo aunque se han implicado desórdenes en distintas rutas celulares entre las que se incluyen el procesamiento génico, la proteostasis y la agregación proteica, el estrés oxidativo o las alteraciones en el microambiente neuronal. En ellas, el desarrollo de enfermedad probablemente se deba a la interacción de agentes ambientales en sujetos genéticamente predispuestos. Futuros estudios de investigación contribuirán al conocimiento patogénico de la ELA, lo que permitirá el planteamiento de nuevas estrategias terapéuticas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

