







Alpha-1 antitrypsin deficiency (AATD) is a rare hereditary condition caused by decreased plasma and tissue levels of alpha-1 antitrypsin (AAT) that can lead to serious lung and liver disease in children and adults. AATD patients face challenges such as under diagnosis, clinical variability, and limited treatment options for liver disease. Early detection and biomarkers for predicting outcomes are needed to improve patient outcome. Currently, the only approved pharmacological therapy is augmentation therapy, which can delay the progression of emphysema. However, alternative strategies such as gene therapy, induced pluripotent stem cells, and prevention of AAT polymerization inside hepatocytes are being investigated. This review aims to summarize and update current knowledge on AATD, identify areas of controversy, and formulate questions for further research.
El déficit de alfa-1 antitripsina (DAAT) es una enfermedad hereditaria poco frecuente causada por la disminución de los niveles plasmáticos y tisulares de alfa-1 antitripsina (AAT) que puede provocar enfermedades pulmonares y hepáticas graves en niños y adultos. Aquellos con DAAT se enfrentan a retos como el infradiagnóstico, la variabilidad clínica y a las limitadas opciones de tratamiento para la enfermedad hepática. La detección precoz y los biomarcadores para predecir los resultados clínicos son necesarios para mejorar la evolución de los pacientes. En la actualidad, el único tratamiento farmacológico aprobado es la terapia de reposición, que puede retrasar la progresión del enfisema. Sin embargo, se están investigando estrategias alternativas como la terapia génica, las células madre pluripotentes inducidas y la prevención de la polimerización de la AAT en el interior de los hepatocitos. Esta revisión pretende resumir y actualizar los conocimientos actuales sobre la AATD, identificar las áreas de controversia y formular preguntas para futuras investigaciones.
Alpha-1 antitrypsin deficiency (AATD) is a rare inherited condition (ORPHA:60) characterized by reduced plasma levels of alpha-1 antitrypsin (AAT), a 52 KDa serine-protease inhibitor glycoprotein encoded by the gen SERPINA1 located on chromosome 14, synthesized and secreted mainly by hepatocytes (and to a lesser extent by monocytes, macrophages, neutrophils, and pulmonary epithelial cells). AAT's main function is to protect the lungs from damage caused by proteolytic enzymes such as neutrophil elastase (NE), cathepsin G, and proteinase 3, released by activated neutrophils during inflammatory or infectious processes. By neutralizing the excess of proteases, AAT helps prevent excessive elastin and collagen IV degradation in the lung's connective tissue.1,2
Over 500 mutations have been described at the SERPINA1 locus.3 The most frequent deficient variants are the Z (Glu342Lys) and S alleles (Glu264Val), while the wild-type is referred to as the M allele. The presence of the Z allele results in the misfolding and polymerization of the AAT, leading to its accumulation (about 70% of all AAT produced) in the hepatocytes’ endoplasmic reticulum (ER), which may result in chronic liver disease (fibrosis, cirrhosis, and liver cancer).4 Autophagy plays a role in the partial degradation of these polymers, leading to the production of inclusion bodies that can be identified through positive acid-Schiff staining and diastase resistance, a unique characteristic of the disease. Approximately 15% of the AAT is released into the bloodstream in polymer form, while an additional 15% is secreted in a functional state, albeit with a significantly reduced ability to inhibit NE.5 Mutations that promote the orderly polymerization of the protein, like the Z allele, can cause an ER overload response, a reaction that involves the calcium-dependent nuclear factor (NF)-κB signaling and elicits a pro-inflammatory response.6
The S mutation does form polymers but at a lower rate than the Z mutation resulting in reduced hepatocyte retention and the absence of liver disease. Although the misfolded S protein is eliminated through the ER-associated protein degradation system and the unfolded protein response and by autophagy, a percentage is properly folded and secreted into the circulation leading to intermediate plasma levels.1,2
As a consequence of these mutations, individuals with AATD may experience reduced levels of AAT in the bloodstream and tissues (particularly the lungs), resulting in a diminished ability to inhibit NE, making the lungs susceptible to proteases and increasing the risk of emphysema, chronic bronchitis, bronchiectasis, and bronchial asthma. Exposure to smoking, dust, and pollution in occupational settings can exacerbate these risks.1,2 Other conditions that may be linked to AATD include neutrophilic panniculitis and granulomatous polyangiitis, although they are much less prevalent.1
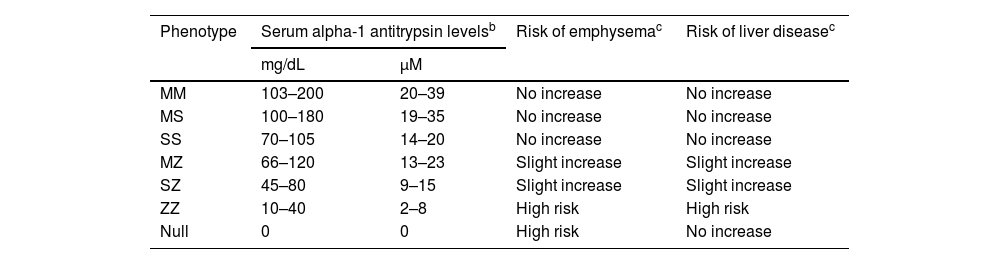
The relationship between AAT plasma level, phenotype, and risk of developing lung and/or liver disease is shown in Table 1.7
Relationship between plasma alpha-1 antitrypsin levels, phenotypes, and associated risk of developing lung or liver disease.a
| Phenotype | Serum alpha-1 antitrypsin levelsb | Risk of emphysemac | Risk of liver diseasec | |
|---|---|---|---|---|
| mg/dL | μM | |||
| MM | 103–200 | 20–39 | No increase | No increase |
| MS | 100–180 | 19–35 | No increase | No increase |
| SS | 70–105 | 14–20 | No increase | No increase |
| MZ | 66–120 | 13–23 | Slight increase | Slight increase |
| SZ | 45–80 | 9–15 | Slight increase | Slight increase |
| ZZ | 10–40 | 2–8 | High risk | High risk |
| Null | 0 | 0 | High risk | No increase |
In addition to its antiprotease activity, AAT has anti-inflammatory and immunoregulatory properties. AATD is increasingly recognized as an inflammatory disease in which neutrophils play a crucial role in the associated inflammation. Several studies have revealed that AATD patients have a significantly higher number of neutrophils in their lungs than healthy individuals, which, together with the low AAT levels, could contribute to the lung damage observed in some patients.8,9
AATD patients face three major challenges: (i) underdiagnosis, (ii) a high degree of clinical variability, and (iii) the lack of specific treatment options for liver disease. It is estimated that about 200,000 and 1.5 million individuals have the PIZZ and PISZ phenotypes, respectively, of which more than 90% remain undiagnosed. Since the early symptoms of the disease are quite similar to those of other respiratory pathologies, the initial clinical diagnosis is challenging, particularly in neonates and children. In addition, under diagnosis leads to an average diagnostic delay of 5–10 years, which may negatively affect the prognosis of patients.10–12
The variance in symptom severity and potential outcomes among those patients diagnosed with AATD is significant, with neither serum AAT levels nor phenotype providing precise predictions regarding the development of severe pulmonary or liver disease. This observation suggests that additional factors, including genetics, epigenetics, environment, and lifestyle, may contribute to the disease's severity. Consequently, it is imperative to adopt innovative approaches to detect AATD earlier and identify new prognostic biomarkers, thus improving patient outcomes and reducing morbidity and mortality.13
The only approved pharmacological therapy to specifically treat severe pulmonary disease in AATD patients is augmentative therapy. Implementing this therapeutic approach has generated significant debate and discussion within the field. However, no pharmacological intervention currently exists for treating AATD-associated liver disease. Researchers are exploring novel therapeutic avenues such as gene therapy, induced pluripotent stem cells, and gene silencing strategies to prevent AAT polymerization within hepatocytes.13
Upon analysis of the provided information, it is apparent that the realm of AATD is undergoing a rapid evolution with a plethora of novel and intriguing discoveries. This review aims to summarize and update current knowledge on AATD, identify areas of controversy, and formulate questions for further research.
Alpha-1 antitrypsin deficiency in childhoodHomozygous AATD individuals for the Z allele (PIZZ) do not typically receive a diagnosis of emphysema in their childhood, although they may experience an increased likelihood of asthma development. However, the onset of emphysema and asthma may both emerge in their adult lives. As mentioned before, exposure to environmental pollutants, lung exposure in occupational settings, and smoking can all contribute to irreversible and progressive lung disease. It has been reported that exposure to second-hand smoke and environmental air pollutants during childhood is a significant risk factor for developing adult AATD-associated emphysema.14,15
Recurrent respiratory manifestations may be an exacerbating factor in the progression of an underlying respiratory problem. Therefore, pediatricians should aim to prevent respiratory infections and efficiently manage bronchial hyper-reactivity in their patients. A highly effective approach to achieving this objective is through the timely administration of appropriate vaccines as per the child's age, including but not limited to hepatitis A and B, pneumococcal 13-valent vaccines, and annual influenza vaccines.1 Ideally, PIZZ AATD children should receive a baseline assessment from an adult pulmonologist upon age 18 unless they exhibit respiratory symptoms that necessitate treatment during childhood.14
The progression of AATD-related liver disease is characterized by a significant level of variability among individuals, with no clear comprehension as to why some individuals are more prone to the disease than others. The majority of PIZZ children are asymptomatic and recover during their early childhood. However, a fraction of them (ranging from 10% to 50%) may present with hepatic abnormalities such as elevated liver enzymes, neonatal hepatitis, enlarged liver, and nutritional deficiencies that may persist into childhood. Blood tests may reveal high levels of bilirubin and transaminases, decreased albumin levels, or coagulopathy, which can be attributed to a vitamin K deficiency or liver dysfunction.1,14,15
A comprehensive and unbiased research investigation was undertaken by Sveger et al. during the 1970s to examine AATD's natural history. A screening was conducted on 200.000 newborns in Sweden, from which 127 individuals with the PIZZ genotype were identified and closely monitored as a study cohort. The study findings revealed that while life-threatening liver disease could manifest in the early stages of life, about 80% of PIZZ patients who presented with neonatal cholestasis remained healthy and free from chronic illness by age 18. The incidence of life-threatening liver disease during childhood was estimated to be approximately 5%, with only 2–3% of children requiring transplantation due to fibrosis or cirrhosis.14–16 More recently, findings from a newborn screening program have also revealed a low rate of life-threatening childhood liver disease.15 A recent systematic review of literature aimed to clarify the clinical course of AATD in both children and adults, as well as evaluate the efficacy of liver transplantation, revealed that 7.5% of children developed liver cirrhosis, 9% showed abnormal liver function tests, 6.9% exhibited portal hypertension, 1.9% presented jaundice, and 16.5% required liver transplantation. No cases of hepatocellular carcinoma were reported. Mortality rates ranged from 0% to 25.5%, significantly improving since the 1980s when liver transplantation became standard practice. Outcomes following liver transplantation were highly favorable, with no recurrence of liver disease or pulmonary complications, suggesting its effectiveness in treating liver disease resulting from AATD.17
Altogether these findings have provided crucial insights into the natural progression of AATD and have the potential for clinical decision-making and patient management strategies. Pediatricians should be aware of patients with persistent unconjugated hyperbilirubinemia, elevated transaminases, neonatal hepatitis syndrome, or other indications of hepatic impairment. Furthermore, careful evaluation is required for older children exhibiting chronic liver disease, cirrhosis, or portal hypertension symptoms. Lastly, pediatricians must consider the medical history of children whose progenitors have been diagnosed with AATD (Table 2).1,17
Candidates for determination of plasma alpha-1 antitrypsin levels in childhood1
| 1 | Infants with: |
| • Persistent unconjugated hyperbilirubinemia | |
| • Elevated transaminases | |
| • Neonatal hepatitis syndrome | |
| 2 | Older children with: |
| • Chronic liver disease | |
| • Cirrhosis | |
| • Portal hypertension | |
| 3 | Children of patients with AATD (family testing) |
AATD is frequently underdiagnosed and requires attention and screening in particular patient groups. Patients who suffer from Chronic Obstructive Pulmonary Disease (COPD), unexplained liver disease, unresponsive asthma, c-ANCA vasculitis, necrotizing panniculitis, granulomatosis with polyangiitis or bronchiectasis, as well as first-degree relatives of individuals with AATD, should be considered for testing (Table 3).18
Candidates for determination of plasma alpha-1 antitrypsin levels.a
| 1 | Chronic obstructive pulmonary disease (COPD) |
| 2 | Adults with bronchiectasis |
| 3 | Partially reversible adult asthma |
| 4 | Consanguineous relatives of alpha-1 antitrypsin deficient patients |
| 5 | Clinic of dyspnea and chronic cough in many members of a family |
| 6 | Hepatopathy of unknown cause |
| 7 | Absence of alpha-1 peak on proteinogram |
| 8 | Panniculitis or vasculitis of unknown cause |
Despite these recommendations, AATD is still widely unrecognized, with fewer than 10% of affected individuals being clinically diagnosed. Patients are often subjected to substantial delays in diagnosis, with some patients even waiting up to 5.6 years and seeking medical attention from several clinicians before receiving a definitive diagnosis. These observations highlight the pressing need for increased awareness and improved diagnostic measures for AATD.1
According to current recommendations, the AATD testing process should be initiated with the assessment of serum AAT levels measured by nephelometry in those individuals fulfilling the criteria shown in Table 3. Additionally, it is of utmost importance to evaluate the C-reactive protein level, as AAT can potentially increase during infection or inflammation. A serum level equal to or exceeding 1.1g per liter and a C-reactive protein level within the normal range signifies a normal AAT status. Conversely, if the serum level is identified as less than 1.1g per liter or noteworthy clinical concerns arise, it is advisable to request phenotyping or genotyping from a specialized laboratory. In cases where the results are inconclusive, gene sequencing should be performed. In any case, it is recommended that patients be referred to a center that specializes in AATD for further evaluation. Certain guidelines suggest concurrent evaluation of AAT levels and genotyping for optimal results.5 Although no universal diagnostic algorithm exists, a tentative algorithm for AATD diagnosis is shown in Fig. 1.19

Algorithm for the diagnosis of alpha-1 antitrypsin deficiency. Adapted from Jardim et al.19
The severity of AAT symptoms and prognosis can vary greatly among patients, and neither their serum AAT levels nor phenotype can effectively identify those patients who will develop severe pulmonary or liver disease. Clinical data suggest that other factors, such as genetics, epigenetics, environment, and lifestyle, may also contribute to the severity of the disease. Therefore, new strategies are necessary to enable early detection of AAT and the discovery of new prognostic biomarkers to reduce morbidity and mortality for those with the condition.2
AATD prognosis is heavily influenced by respiratory disease. The most common form of respiratory disease is early-onset emphysema, which affects 58%–72% of patients.20,21 It is crucial to note that smoking can significantly exacerbate lung disease and is the primary risk factor for developing rapidly progressive COPD in individuals with AATD. Epidemiological studies have conclusively proven that AATD individuals who continue smoking are at a higher risk of developing emphysema, increased airflow obstruction, lower carbon monoxide diffusing capacity (DLCO), and increased sputum production. Furthermore, current smokers have a much higher annual lung function loss rate than those who have never smoked or quit smoking. A recent study showed that PISZ patients were less susceptible to cigarette smoke than PIZZ patients. Multivariate analysis revealed that PISZ patients were less likely to have emphysema and had better survival than PIZZ patients, given the same level of smoke exposure. However, the lung function decline did not differ significantly.22–24
There has been ongoing debate about the possible link between lung disease and PIMZ individuals for several years. This is a significant concern as PIMZ individuals are common, and even a slight increase in the risk of COPD could have major public health implications. Initially, it was difficult to establish a definitive association between PIMZ and COPD development since studies were performed with a small number of patients. Later studies have shown that PIMZ and PISZ individuals who smoke have an increased risk of COPD, while non-smokers do not. Therefore, it is strongly recommended that PIMZ and PISZ individuals receive an initial diagnosis as soon as possible, followed by intensive counseling to prevent non-smokers from starting smoking or to assist current smokers in quitting. Individuals diagnosed with COPD should undergo regular monitoring using established protocols. However, the need for more frequent check-ups is uncertain due to unclear information regarding lung function decline acceleration in individuals who have quit smoking compared to PIMM carriers with COPD. Further investigation is required to determine the best action for monitoring and treating COPD in these individuals.5,25
Certain uncommon variants of AAT, such as the F, I, and Mmalton alleles, are linked to a higher chance of COPD only when inherited alongside a Z or null allele. Homozygous individuals for the null allele lack any circulating AAT and usually suffer more severe lung disease than PI ZZ or PI SZ allele carriers. However, they are not at a greater risk of liver disease.5
Liver disease linked to AATD can vary in severity and may impact adults, especially those above 50. It can lead to serious conditions such as cirrhosis and hepatocellular carcinoma, requiring a liver transplant. It has been shown that individuals with the PIMZ phenotype have a higher risk of developing liver fibrosis or cirrhosis. However, non-alcoholic steatohepatitis and alcohol consumption may also contribute significantly to liver disease in this group.15
Several studies indicate that around 50% of PI ZZ homozygotes show liver inflammation, with cirrhosis developing in 2%–43% of cases. The risk of developing liver disease also increases with age. Individuals carriers of the PIZZ phenotype should receive proper monitoring for liver disease. Follow-up of asymptomatic patients by liver biopsy is not recommended due to the procedure's invasiveness. Transient elastography is reliable for ruling out advanced fibrosis (stage F3 or F4). However, it may not be as effective for detecting lower levels of fibrosis. Several biomarkers are currently under study, including the aspartate aminotransferase platelet ratio index, the Fibrosis-4 (FIB-4) score, and the γ-glutamyltransferase level. However, cutoff values for liver disease associated with AATD remain to be defined.5,15
Alpha-1 antitrypsin deficiency treatment: the augmentation therapyThe only approved pharmacological treatment for AATD is intravenous augmentation therapy. This involves infusing plasma-purified AAT obtained from healthy individuals into AATD patients. This therapy aims to protect the lungs from the damaging effects of uncontrolled neutrophil elastase, which can slow down the progression of emphysema. Augmentation therapy was granted approval by the Food and Drug Administration in 1987 based on its biochemical efficacy and pharmacokinetics in maintaining protective AAT levels in blood and lung tissue without proof of clinical efficacy. This aspect has therefore been the subject of extensive debate.1,15,26
Through initial studies, the research primarily focused on the decline of forced expiratory volume in one second (FEV1) and mortality rates. Results indicated that the treated group displayed a decrease in FEV1 decline.27–30 Subsequently, further observational studies revealed that AAT augmentation therapy led to a slower decline in FEV1 and a decrease in mortality compared to those not receiving this treatment.1,29,31 However, while the augmentation therapy yielded positive results, the reduction in lung function loss was primarily observed in patients with an FEV1 between 35 and 60%. As such, the recommended course of treatment was only advised for patients who fall within this range of lung function impairment.27,32 Augmentation therapy is not recommended for heterozygous individuals with one deficiency allele and individuals with two deficiency alleles who do not have airflow obstruction.5
Later randomized, controlled trials were undertaken to investigate the primary efficacy outcome of reducing the loss of lung density. Pulmonary function tests and lung density measured by computed tomography (CT) scan were studied in 30 patients using a randomized placebo-controlled clinical trial. No significant differences in the pulmonary function tests were observed but a tendency toward improved lung density (p<0.07) in the treatment versus the placebo group. The study concluded that the decline in FEV1 is not an appropriate method to assess the efficacy of augmentation therapy due to the significant number of patients required.33 Recent studies have explored alternative outcome metrics such as the carbon monoxide diffusing capacity (DLCO) or lung density measured by CT scan. The results from these studies have shown that a decline in DLCO is observed before FEV1 decreases, and both DLCO and lung density demonstrate lung parenchyma loss, even in severe diseases where FEV1 may be stable. Furthermore, lung density assessed by CT correlates with health-related quality of life and is the best predictor of mortality in AATD patients. In the EXACTLE randomized controlled trial, changes in CT lung density in patients receiving AAT augmentation therapy versus placebo were evaluated, and a trend (p=0.068) toward improved lung density was observed. Pooled data from these two clinical trials showed a significant improvement in lung density decline over two years in treated versus untreated patients.34,35
Despite these promising results and the support of multiple meta-analyses, a lack of consensus and an unfavorable Cochrane review indicated that new evidence was needed to establish the potential therapeutic value of augmentative therapy.36,37 To this end, the RAPID clinical trial was conducted. The outcomes of this trial demonstrated that augmentation therapy effectively reduced annual lung tissue loss, as evidenced by a statistically significant reduction of lung density loss measured.38 Moreover, patients who were initially included in the placebo arm of the study agreed to participate in the extension study (the RAPID-OLE clinical trial) and subsequently received augmentation therapy for the next two years displayed a reduction in their lung density decline rate akin to patients initially included in the treatment arm of the study.39
Another subject of debate is the role of replacement therapy in the post-transplant period. The question of whether transplant recipients should receive exogenous AAT still needs to be answered due to insufficient evidence.40 Based on current knowledge, continuing augmentation therapy after lung transplantation may be beneficial, especially for certain patients during the peri-transplant period or if there is a clinical decline. This approach could reduce the risk of post-procedure inflammation and protect the function of the transplanted lungs.40,41
Beyond its clinical efficacy, augmentative therapy can be challenging for both patients and healthcare institutions due to the significant time and resources required. To address these challenges, several patient-centered alternatives are being explored to improve treatment adherence and overall quality of life: (i) a 120mg/kg bi-weekly dosing which has a similar safety profile to a weekly 60mg/kg dose and has been shown to achieve protective serum levels of AAT in ZZ patients. However, the potential long-term impact on the progression of the disease is currently unknown. This therapeutic strategy has benefits such as less exposure to hospital-acquired infections, convenience for full-time workers, lower travel costs, and longer leave periods; (ii) self-administration, a safe and cost-effective option already used in other medical conditions, is being explored in AATD, with patients having reported high satisfaction levels and improved independence. However, the training required for self-administration could be an issue. Other therapeutic options, such as home therapy and the upcoming 4–5-g presentation, are also being explored, although they are only available in some countries.40
There is no approved pharmacological treatment for AATD-associated liver disease, except liver transplant for those patients in advanced stages of the disease. AATD-associated liver disease may be worsened by alcohol and excessive fat consumption. To promote healthy liver function, those individuals with normal liver function should maintain a normal body mass index and avoid alcohol intake.5,15,40
Biomarkers for alpha-1 antitrypsin deficiencySeveral biomarkers that can be reliable indicators of healthy lung or liver function, disease progression, and response to augmentation therapy are being investigated.42
Circulating polymers are being studied as a diagnostic method and possible biomarkers for AATD. Early data shows they may contribute to lung function deterioration, but further research is needed to determine their stability and effectiveness.43,44
Desmosine and isodesmosine are linked to lung elastin degradation and the development of COPD. Levels of these biomarkers are elevated in the biofluids of COPD patients, with or without AATD.45 AAT augmentation therapy may help reduce desmosine excretion in AATD patients.46
Fibrinogen levels are linked to the number of exacerbations, disease severity, and mortality rates in COPD patients. In addition, levels of a particular fibrinogen degradation product (Aa-Val360) increase in patients with AATD, indicating the severity of airflow obstruction, and decrease in those patients receiving AAT augmentation therapy. These findings suggest that Aa-Val360 may be a valuable marker for disease activity in patients with early-stage COPD who may need therapeutic intervention.47
Emerging therapeutic strategiesWith the limited scope of eligible individuals and the limited effectiveness of augmentation therapy, there has been a growing interest in exploring new therapeutic approaches. Several strategies are being investigated, including gene therapy, induced pluripotent stem cells (iPSCs), gene-editing cell-based therapy, and small molecule therapies targeting AAT polymerization within hepatocytes. Additionally, autophagy-enhancing drugs and silencing RNA strategies are being considered as potential therapeutic options.1
Gene therapies have been promising therapeutic strategies for many years.48 Replacement therapies have been studied using both viral48 and non-viral methods.49,50 Initial gene therapies utilizing murine retroviruses demonstrated some efficacy in elevating serum AAT levels but have faced challenges due to immune-related adverse effects or diminishing effectiveness.51,52 Second-generation gene therapies using human lentiviruses have shown promise in sustaining AAT increases but have only undergone testing in mouse models and have yet to be translated to human studies. Initially, these methods were thought to only work for emphysema. Still, recent research on AATD mice has shown that it is possible to simultaneously eliminate Z gene expression and introduce the wild-type AAT gene. This approach resulted in high levels of human AAT therapy and a noticeable decrease in the accumulation of Z protein in the liver. However, it's important to note that while this reduction was significant, it wasn’t enough to prevent liver fibrosis.53
The recent advent of efficient genome editing based on the CRISPR/Cas9 system has opened up new strategies for definitive gene correction of the Z-AAT mutation in hepatocytes, which are currently under investigation. Researchers have successfully corrected Z allele mutations in both in vitro and in vivo iPSCs using CRISPR/Cas9. The study aimed to modify iPSCs, making them suitable for engraftment without causing any damage to their DNA. After correcting the Z allele mutation, the results showed a significant reduction in abnormal AAT accumulation and endoplasmic reticulum stress in iPSC-derived hepatocytes.54,55 Despite its potential benefits, this therapy also poses risks, such as the possibility of harmful point mutations and uncertain epigenetic changes, which precludes its use in the clinical practice. While gene-editing techniques show promising therapeutic potential, some important questions still need to be addressed before they can be used in clinical settings. Specifically, further investigation is required on delivering specifically to hepatocytes and optimizing gene editing efficiencies to achieve the desired physiological effects. Additionally, there is a need to prevent off-targeted mutagenesis, which has been recently reported.54
A set of small molecules has been discovered that can limit the formation of Z allele AAT polymers and promote the secretion of AAT in AATD iPSCs. These molecules, along with intrabodies, have the potential to stabilize the polymerization process. Researchers have found a peptide that can effectively stop the polymerization of a mutated AAT-Z protein by targeting a particular hydrophobic region. However, this peptide does not prevent the protein's secretion. Instead, it promotes its degradation within the cell.56,57
Lately, there has been an increasing interest in using autophagy enhancement as an alternative treatment for liver transplantation. Two drugs, carbamazepine and rapamycin, have been found to improve autophagy, which can lead to the degradation of misfolded Z-AAT and a reduction in hepatic fibrosis in a mouse model of AATD liver disease. Currently, carbamazepine is being evaluated in a double-blind, placebo-controlled, randomized clinical trial for AATD-associated severe liver disease.5,58,59
Additionally, another approach involves using interference RNA (RNAi) to silence Z-AAT in hepatocytes. Preclinical data have shown that this method can decrease the formation of inclusion bodies and hepatic damage in a mouse model of the disease.60
ConclusionsAATD is a medical condition frequently not diagnosed in its early stages. This can lead to a delay in treatment that may last for years and negatively affect the prognosis of the disease. It is, therefore, crucial to increase awareness of this condition and ensure prompt diagnosis and treatment to enhance patient outcomes. This aspect is especially important because research supports the clinical efficacy of augmentation therapy, and the new upcoming therapies for treating the disease being studied show promising results. Identifying relevant biomarkers is crucial for accurately predicting disease progression and assessing the effectiveness of treatment. This will enable healthcare professionals to categorize patients effectively, resulting in optimal patient outcomes and informed treatment decisions.
Ethical considerationsWritten informed consent was not requested as no patient information was used in the writing of this review article.
FundingThis work has been funded by the Valencian Society of Pneumology 2021, the ISCIII #PI17/01250, and the European Regional Development Funds (ERDF).
Conflict of interestFrancisco Dasí has received grants for registration and attendance to congresses from CSL Behring and Grifols S.A.
We want to show our deepest gratitude to the Spanish Association of Patients with alpha-1 antitrypsin deficiency for donating research funds to our research group on rare respiratory diseases at IIS INCLIVA/UVEG.

