







Este trabajo tiene como objetivo dar respuesta a preguntas que con frecuencia se hacen médicos que atienden a pacientes con enfermedad de Wilson (EW) o pacientes en quienes se plantea la sospecha diagnóstica de que se trate de una EW. Tiene 2 partes, una con respuestas a preguntas relacionadas con el diagnóstico de la enfermedad y otra con respuestas a preguntas relacionadas con el tratamiento. Al final se incluye un breve apéndice con respuestas a preguntas que no se pueden incluir en las 2 anteriores categorías.
The present article aims to provide answers to questions frequently asked by physicians attending patients with Wilson's disease (WD) or those with a suspected diagnosis of WD. The article is divided into 2 parts: a first part with answers to questions relating to the diagnosis of this entity and a second with answers to questions concerning treatment. A brief appendix is included with responses to questions not falling into either of these 2 categories.
La enfermedad de Wilson (EW) es una enfermedad autosómica recesiva caracterizada por una acumulación progresiva de cobre en el organismo, debido a un fallo en la excreción biliar del este metal y en su incorporación a la apoceruloplasmina para formar ceruloplasmina1. Estos defectos son la consecuencia de una alteración estructural y funcional de la proteína transportadora del cobre, localizada en el citoplasma de los hepatocitos, causada por la mutación en los 2 alelos del gen ATP7B, ya sea en forma homocigota o heterocigota compuesta2-4. El cobre acumulado causa daño en el hígado y en el cerebro.
Diagnóstico de la enfermedad de WilsonDebe pensarse en el diagnóstico de EW en cualquier persona con alteraciones hepáticas sin causa reconocida, especialmente en personas jóvenes, y en las que simultáneamente presenten alteraciones del movimiento. No existe ninguna prueba específica para el diagnóstico de EW más que la prueba genética, cuando se encuentra una mutación homocigota o una heterocigosis compuesta del gen ATB7B. En la práctica el diagnóstico se basa en la combinación de manifestaciones clínicas y pruebas de laboratorio (ceruloplasmina baja y cupruria elevada). Ninguna de ellas individualmente permite confirmar o excluir el diagnóstico.
En una reunión de expertos en Leipzig en 20015 se propuso un sistema de puntuación que ha facilitado notablemente el diagnóstico de EW (tabla 1). Este sistema ha sido validado en la población adulta6,7, pero no en niños8. A pesar de ello, en la práctica clínica se plantean a menudo problemas de interpretación que se intentan resolver en este artículo.
Sistema de puntuación para el diagnóstico de enfermedad de Wilsona
| Anillo de Kayser-Fleischer | |
| Presente | 2 |
| Ausente | 0 |
| Síntomas neurológicos | |
| Graves | 2 |
| Ligeros | 1 |
| Ausentes | 0 |
| Ceruloplasmina | |
| Normal (> 20mg/dl) | 0 |
| 10-20 | 1 |
| < 10 | 2 |
| Cupruria | |
| Normal | 0 |
| 1-2 × N | 1 |
| > 2 × N | 2 |
| Cobre intrahepático | |
| Normal (< 50 μg/g) | 0 |
| Entre 50 y 250 | 1 |
| > 250 | 2 |
| Anemia hemolítica Coombs negativa | |
| Ausente | 0 |
| Presente | 1 |
| Estudio molecular | |
| Mutación en 2 cromosomas | 4 |
| Mutación en 1 cromosoma | 1 |
| Ausencia de mutación | 0 |
Puntos totales: 4 o más, diagnóstico de EW; 3 puntos, diagnóstico posible; 2 puntos o menos, diagnóstico de EW improbable.
Disponemos de quelantes del cobre, la d-penicilamina y la trientina9,10, que son promotores de la excreción renal de cobre, y de un bloqueador de la absorción intestinal del cobre, la sales de cinc11. El tratamiento con cinc determina por una parte un balance negativo de cobre de<1mg/día y también estimula la producción citoplasmática de metalotioneína, proteína que fija el cobre en el citoplasma de los hepatocitos en forma no tóxica y actúa por tanto como un quelante endógeno. El empleo de un cuarto fármaco, el tetratiomolibdato, utilizado por los veterinarios para tratar la intoxicación por cobre de los animales, podría ser adecuado para las formas de expresión neurológica, pero su comercialización todavía no ha sido autorizada12. Los quelantes del cobre son productos con más afinidad para el cobre que las proteínas que lo transportan. La d-penicilamina fue el primer fármaco utilizado en esta enfermedad13 y el más potente, pero presenta efectos secundarios (tabla 2), algunos de ellos muy severos, que obligan a su sustitución.
Efectos adversos de la d-penicilamina
| Precoces (primeras 2 semanas) |
| Reacción de hipersensibilidad (fiebre, erupción cutánea, adenopatías, proteinuria) |
| Toxicidad medula ósea (anemia, trombocitopenia, neutropenia) |
| Tardías |
| Nefrotoxicidad (proteinuria) |
| Síndrome de Goodpasture |
| Elastosis perforans |
| Seudolupus |
| Liquen plano |
| Disgeusia |
| Macromastia |
| Daño pulmonar |
La trientina fue introducida en 1969 por Walshe10, el mismo que descubrió la utilidad de la penicilamina, para tratar a un paciente que no toleró la penicilamina. Tiene menos efectos adversos que esta última y similar eficacia.
Las sales de cinc fueron utilizadas por primera vez en la EW por investigadores holandeses11. Carecen de efectos secundarios importantes, pero poseen menor eficacia que los agentes quelantes. Se pueden prescribir como acetato o como sulfato de cinc, preparados de forma oficinal o como Wilzin®, que es un preparado comercial que contiene acetato de cinc.
Para el tratamiento de las formas neurológicas se ha explorado la utilidad de una molécula, el tetratiomolibdato, utilizado con éxito por los veterinarios para el tratamiento de la intoxicación por cobre en los animales, pero todavía no ha sido autorizado su empleo en humanos12.
El tratamiento debe mantenerse toda la vida, y en la mayoría de pacientes determina una respuesta satisfactoria. La supervivencia de la gran mayoría de pacientes tratados coincide con la de la población general13. En los pacientes descompensados con fallo hepático fulminante el tratamiento es el trasplante hepático.
Dudas y preguntas frecuentesEste trabajo tiene como objetivo dar respuesta a preguntas que con frecuencia se hacen médicos que atienden pacientes con EW o pacientes en quienes se plantea la sospecha diagnóstica de que se trate de una EW. Tiene 2 partes, una con respuestas a preguntas relacionadas con el diagnóstico de la enfermedad y otra con respuestas a preguntas relacionadas con el tratamiento. Al final se incluye un breve apéndice con respuestas a preguntas que no se pueden incluir en las dos anteriores categorías.
Preguntas relacionadas con el diagnóstico¿La ceruloplasmina en suero baja indica siempre enfermedad de Wilson?No, ya que también tienen valores discretamente bajos el 10-20% de los sujetos heterocigotos para el defecto genético de la EW.
También es baja en la aceruloplasminemia, enfermedad muy rara caracterizada por la asociación de diabetes, degeneración retiniana, distonía, disartria y ferritina en sangre elevada14. También, aunque muy infrecuentemente, puede estar levemente descendida en las hepatopatías avanzadas por déficit de síntesis o en casos de malabsorción. No obstante, valores inferiores a 10mg/dl señalan fuertemente que se trate de una EW.
¿La ceruloplasmina normal excluye el diagnóstico de enfermedad de Wilson?No, si la determinación se ha hecho por el método de la inmunoprecipitación, que detecta a la vez ceruloplasmina y apoceruloplasmina. Tiene más fiabilidad la determinación de ceruloplasmina por el método enzimático que mide la actividad oxidasa de la ceruloplasmina15,16. No obstante, incluso con esta prueba el 15% de pacientes con EW tienen valores de ceruloplasmina en el rango bajo de la normalidad. Los valores normales oscilan entre 20 y 40mg/dl.
En algunos pacientes con EW asintomática y sin disfunción hepática se alternan resultados normales y valores ligeramente bajos, si se efectúan exámenes seriados. También se encuentran valores normales de ceruloplasmina en mujeres con EW que toman contraceptivos orales o están embarazadas.
Por consiguiente, si la hipótesis diagnóstica de EW es fuerte en un determinado paciente, pero la ceruloplasmina no es baja deben valorarse otros exámenes como la cupruria y el cobre plasmático libre, y eventualmente medir la concentración hepática de cobre, antes de descartar este diagnóstico.
¿Cuál es el cut-off de la cupruria de 24h para distinguir un valor normal de uno patológico?El cobre de orina procede del cobre plasmático no unido a la ceruloplasmina. El examen de la cupruria de 24h es muy útil ya que los pacientes con EW excretan más cobre que los sujetos sanos. Tienen cobre libre en exceso, pues en general tienen poca ceruloplasmina. En los niños debe considerarse sospechoso de EW un valor por encima de 40 μg8,17, mientras que en los pacientes adultos es preferible utilizar un cut-off de 100 μg. La cupruria es más elevada en pacientes con enfermedad avanzada que en los que están asintomáticos.
Es una prueba de difícil valoración porque requiere una buena recogida de la orina, y que la muestra sea de exactamente 24 h, pero no más ni menos.
¿Es útil la prueba de excreción de cobre urinario después de penicilamina?Esta prueba consiste en la medición de la cupruria de 24 h el mismo día en que el paciente toma 1.000mg de d-penicilamina con 12 h de intervalo (500mg por la mañana y 500 por la noche), recogiendo orina a partir de la toma de la primera dosis.
Aunque en niños el estudio del King's College Hospital mostró que una cupruria>1.690 μg/24 h es diagnóstica de EW18, en estudios posteriores se observaron valores de excreción forzada de cobre urinario en niños con EW confirmada mucho más bajos19,20. En adultos la prueba no está validada, y da un resultado normal en muchos pacientes asintomáticos7, por lo que no parece necesario efectuarla17.
¿Debe medirse la cupremia?Sí, pero solo tiene interés la determinación del cobre libre, que está elevado en los pacientes con EW como consecuencia del paso a la sangre del cobre de los hepatocitos no unido a la ceruloplasmina. La cupremia total refleja mayoritariamente el cobre de la ceruloplasmina. Por tanto en los pacientes con EW con ceruloplasmina muy baja nos encontramos con una cupremia total baja, aunque el cobre libre puede estar elevado.
Para calcular el cobre libre se ha se restar del cobre plasmático total (en μg/dl) el valor de la ceruloplasmina (medida por el método enzimático) multiplicado por 3, ya que cada molécula de ceruloplasmina aporta 3 μg de cobre. La cupremia libre normal es<10 μg/dl. Los pacientes con EW presintomáticos tienen entre 10 y 20 μg/dl y los pacientes sintomáticos>20 μg/dl. Debe tenerse se en cuenta que la determinación de la ceruloplasmina debe haberse efectuado por el método enzimático, ya que el inmunológico sobreestima la cantidad de ceruloplasmina, ya que también detecta la apoceruloplasmina1. La concentración de cobre libre se puede calcular fácilmente en la siguiente página web: http://www.wilsonsdisease.org/wilson-disease-patients/wilsondisease-calculator.php
La medición del cobre libre tiene también utilidad en el seguimiento de los pacientes tratados, ya que el tratamiento se considera eficaz si es capaz de reducir los valores de cupremia libre a unos 10 μg/dl.
¿Qué utilidad tiene el examen del anillo de Kayser-Fleischer?Con relación a la utilidad diagnóstica del anillo de Kayser-Fleischer hay que tener en cuenta los siguientes conceptos:
- a)
Se halla presente en el 90% de los casos neurológicos, pero solo en la mitad de los pacientes hepáticos, por lo que la ausencia del anillo no excluye el diagnóstico de EW20,21.
- b)
En los casos con anillo es útil examinar al paciente a los 6 meses de iniciado el tratamiento de la EW para comprobar si ha desaparecido, lo que indicaría eficacia del tratamiento22.
- c)
Se han descrito raros casos de anillo de Kayser-Fleischer en pacientes con colestasis crónica23, ya que en esta situación también se retiene el cobre que no puede ser eliminado debido a la obstrucción biliar.
Conviene que el examen lo efectúe un oftalmólogo experto.
¿Qué tipo de síntomas neurológicos obligan a sospechar una enfermedad de Wilson?Las manifestaciones neurológicas de la EW son muy variadas y reflejan una disfunción cerebelosa o extrapiramidal. Las más frecuentes son temblores, intencionales y de reposo, rigidez, ataxia y distonía, varias de las cuales pueden coincidir en el mismo paciente24. Algunos pacientes se quejan de disfagia y de salivación excesiva25. La disartria es una manifestación común y a veces la inicial. No hay anomalías sensitivas. Los temblores y la rigidez pueden inducir a confusión con la enfermedad de Parkinson.
En ocasiones los padres aprecian mal rendimiento escolar o cambios de carácter que pueden corresponder a la primera manifestación de la enfermedad. Se ha descrito un signo característico, aunque infrecuente, que es la micrografía26 y una escritura temblorosa al hacer líneas rectas.
Las lesiones más avanzadas incluyen una rigidez excesiva, ataxia y movimientos coreico-atetóticos.
¿Existe daño hepático en los pacientes con presentación neurológica?Por lo general sí lo hay, aunque en muchos casos subclínico, e incluso algunos no tienen hepatopatóa. Solo la mitad de ellos tienen cirrosis hepática.
¿Cuándo hay que hacer una biopsia hepática ante una sospecha de enfermedad de Wilson?Cuando hay una sospecha de la enfermedad motivada por hallazgos clínicos o bioquímicos, pero no la seguridad diagnóstica, la biopsia hepática es útil, aunque no exista ningún cambio histológico específico de EW27, porque permite cuantificar el cobre hepático.
El examen histológico puede inducir la sospecha de EW cuando se detecta una esteatosis macrovacuolar en un niño o una persona joven, sin causa que la justifique, como consumo de alcohol, sobrepeso, diabetes o dislipidemia, o cambios de hepatitis crónica sin virus ni alteraciones de la inmunidad. Es conveniente en estos casos hacer una tinción de orceína, con rubeánico o con rodanina, aunque son negativas en muchos casos de EW, especialmente en las formas iniciales de enfermedad28,29. La sensibilidad de las tinciones histoquímicas aumenta si las secciones parafinadas de la biopsia hepática se dejan 24h en xilol, en lugar de los 10min habituales30. La orceína tiñe un pigmento de color negro distribuido irregularmente en los hepatocitos formado por metalotioneína, que es la proteína a la que está fijado el cobre intrahepatocitario. La rodanina y el ácido rubeánico tiñen el cobre, que se ve como un pigmento de color rojo con la primera tinción y de color negro con la segunda.
La biopsia hepática es indispensable para efectuar la cuantificación del cobre intrahepático mediante espectrofotometría de absorción atómica28. Si la determinación de cobre en tejido seco supera los 250 μg/g el diagnóstico de EW es seguro si se han descartado otras causas de acumulación hepática de cobre como las colestasis crónicas y la cirrosis infantil de la India27. Si es inferior a 50 μg/g, que es el límite superior de la normalidad, el diagnóstico de EW es muy improbable. Si el cobre se halla entre 50 y 250 μg/g de tejido hepático seco el diagnóstico de EW es probable31. Ferenci et al.31 han propuesto bajar a 75 μg/g el umbral para la concentración de cobre con valor diagnóstico para EW. Valores superiores a los normales, pero inferiores a 250 μg/g en algunos casos con EW, se pueden explicar porque el depósito de cobre acumulado en el hígado no es homogéneo, o porque haya mucho tejido fibroso en el espécimen de biopsia examinado, ya que el tejido fibroso no contiene cobre.
Es importante que el fragmento de biopsia que se destine a la determinación del cobre intrahepático tenga una longitud>1cm, con objeto de no subestimar el valor obtenido. Para la medición del cobre intrahepático se puede utilizar el bloque de parafina del que se han obtenido los cortes para el examen histológico. No es necesario hacer una segunda biopsia para medir la concentración de cobre en tejido fresco.
¿Cuándo no hay que hacer una biopsia hepática?No debe hacerse en los pacientes ya diagnosticados porque la biopsia no aportará información útil. Tampoco en los que están en tratamiento, ya que en la mayoría de biopsias hepáticas de pacientes tratados sigue habiendo elevadas concentraciones de cobre, aunque estén con pruebas hepáticas normales32. Esto es debido a que tanto los quelantes como el cinc estimulan la producción de metalotioneína que fija el cobre citoplasmático en un estado atóxico, pero no eliminan el cobre en exceso.
¿Cuándo sospechar una enfermedad de Wilson en un paciente ya diagnosticado de alguna enfermedad hepática?Existen pacientes que presentan una EW asociada a una enfermedad hepática de otra etiología33. Si el paciente no presenta clínica neurológica la sospecha diagnóstica solo puede hacerse si se detecta una ceruloplasmina baja o si en la biopsia hepática se aprecian cambios histológicos que son comunes en la EW, como grasa y degeneración glucogénica nuclear. También debe considerarse el diagnóstico de EW en pacientes con hepatitis autoinmune que no respondan al tratamiento inmunosupresor34. En estos casos las dudas diagnósticas se resolverán mediante la determinación de la concentración de cobre en tejido y mediante el análisis genético.
¿Se ha de pensar en enfermedad de Wilson en pacientes de más de 40 años?Siempre que no se haya reconocido la etiología de la enfermedad hepática crónica que presente un paciente de más de 40 años se debe examinar la ceruloplasmina y la cupruria. Si alguna de ellas es anormal es prudente efectuar una biopsia hepática para medir la concentración hepática de cobre. Se ha diagnosticado EW en pacientes septuagenarios35.
¿Cuándo hay que pedir el examen genético?El examen genético es poco útil como examen de rutina, porque es laborioso, caro y no permite en todos los pacientes reconocer una mutación en los 2 alelos. La mayoría de pacientes con EW son heterocigotos compuestos y en cerca del 17% de los pacientes con diagnóstico seguro de EW no se detecta ninguna mutación36.
Solo se debe efectuar la prueba genética si no se ha conseguido llegar al diagnóstico de seguridad de EW mediante las pruebas clínicas y de laboratorio. En las regiones donde predomina una mutación concreta, conviene hacer inicialmente el examen directo de esta mutación, ya que puede simplificar el trabajo y abaratar el coste37.
¿Cómo interpretar los resultados del test genético?En caso de disponer de un resultado de homocigosis para una mutación del gen ATP7B o la demostración de un estado de heterocigosis compuesta el diagnóstico de EW es seguro4. Cuando solo se detecta una mutación en solo un alelo deberá considerarse que se trata de una EW si existen evidencias de enfermedad hepática o neurológica compatible con este diagnóstico, considerando que en el otro alelo tiene probablemente una mutación no identificada o está localizada en un exón no estudiado, pero en los sujetos asintomáticos y sin signos de enfermedad debe interpretarse que se trata de un sujeto heterocigoto.
Cuando la enfermedad se presenta precozmente las posibilidades de no hallar una mutación en los 2 alelos son escasas20, mucho menores que en los casos en que la enfermedad se manifiesta en la edad adulta38.
¿Sería útil en caso de duda diagnóstica un tratamiento de prueba con penicilamina u otro agente para confirmar el diagnóstico?No es recomendable, puesto que el diagnóstico de EW presupone la necesidad de mantener el tratamiento de por vida. Cuando persisten las dudas diagnósticas a pesar del estudio genético, si no se puede identificar una mutación del gen ATP7B en 2 alelos, debe procederse a efectuar una biopsia hepática para poder medir la concentración de cobre en el tejido hepático.
¿Cuándo pensar que una hepatitis fulminante puede tratarse de una enfermedad de Wilson?El fallo hepático fulminante puede ser la manifestación inicial de una EW que había permanecido latente o aparecer semanas o meses después de que el paciente abandonara el tratamiento que seguía de su EW39. Se da en el 5% de los pacientes con EW y es más frecuente en mujeres. La EW representa el 6-12% de los casos de insuficiencia hepática aguda grave.
El cuadro clínico no se distingue del que se observa en el curso de una infección por virus de la hepatitis o una hepatitis tóxica. Es una complicación grave que se sigue casi invariablemente de la muerte si no se efectúa un trasplante hepático urgente40. Por esta razón es fundamental conseguir el diagnóstico con la máxima prontitud y la máxima seguridad.
Un argumento a favor del diagnóstico de EW en caso de insuficiencia hepática aguda grave es la presencia de una hemólisis Coombs negativa, que no se halla presente en todos los pacientes. Por otra parte, si la paciente estaba embarazada o acaba de parir la anemia podría inducir a confundir una EW con un síndrome HELLP.
La presencia de anillo de Kayser-Fleischer tiene un carácter diagnóstico, pero solo se encuentra en un 50% de los pacientes41, y por otra parte es difícil de valorar en el examen con lámpara de hendidura en un paciente que no colabora.
Es frecuente la constatación de una cifra muy baja de fosfatasa alcalina42.
Los datos con mayor especificidad y sensibilidad para el diagnóstico de una EW fulminante son: un cociente AST/ALT>2,2, que posee una sensibilidad de 94% y una especificidad de 86%, y un cociente fosfatasas alcalinas/bilirrubinemia<4, cuya sensibilidad es del 94% y su sensibilidad del 96%43.
¿Debe incluirse la ecografía abdominal en los exámenes que deban hacerse periódicamente a un paciente con enfermedad de Wilson?Únicamente en los pacientes que ya presentan cirrosis en el diagnóstico, ya que aunque infrecuente, el carcinoma hepatocelular también puede incidir sobre un hígado cirrótico por EW44. En ellos debe aplicarse la misma periodicidad para efectuar los exámenes que permitan detectar precozmente un carcinoma hepatocelular injertado sobre la cirrosis.
Preguntas relacionadas con el tratamiento¿Qué fármaco elegir?La elección del fármaco depende de la forma clínica (fenotipo) de la enfermedad, hepática o neurológica, y en el caso de forma hepática si va con síntomas o sin ellos.
La d-penicilamina (Cupripen®) se recomienda para los pacientes con enfermedad hepática sintomática, porque es el fármaco más potente, y las sales de cinc para los pacientes con enfermedad neurológica y los pacientes asintomáticos, ya que es la que comporta menos riesgos para el paciente45,46 (fig. 1). La elección del cinc (Wilzin®) para el tratamiento de los pacientes neurológicos se justifica por la elevada tasa de empeoramiento de los síntomas neurológicos, con frecuencia irreversible, a las pocas semanas de iniciado el tratamiento con d-penicilamina, mientras que con cinc mejora el 90% de los casos tratados47. Los pacientes que no toleran la penicilamina debido a la aparición de efectos secundarios, que son por lo menos el 25% de los que lo inician, deben ser tratados con trientina. Algunos autores prefieren utilizar la trientina como primera opción en los pacientes con hepatopatía descompensada por su menor tasa de efectos adversos48. La penicilamina puede desencadenar la aparición de manifestaciones neurológicas en pacientes que no las tenían, tratados por enfermedad hepática. La trientina posee menor toxicidad que la penicilamina pero también puede causar un empeoramiento en los pacientes neurológicos49. En los pacientes inicialmente tratados con quelantes se pueden utilizar las sales de cinc como tratamiento de mantenimiento.

Estas recomendaciones no deben ser tomadas de forma axiomática, ya que se han descrito pacientes con enfermedad hepática descompensada que entraron en remisión con tratamiento con cinc50 e inversamente casos tratados con sales de cinc porque estaban en fase asintomática que experimentaron un empeoramiento de la enfermedad51-53.
Desde el punto de vista teórico los pacientes con enfermedad hepática descompensada se podrían beneficiar de atacar a la enfermedad simultáneamente con un agente quelante, que eliminaría cobre, y con cinc, que impediría la absorción intestinal de cobre, y ambos fijarían el cobre en situación atóxica al estimular la producción de metalotioneína que se combinaría con el cobre citoplasmático. Sin embargo, esta pauta se ha usado excepcionalmente, ya que es de difícil aceptación por el paciente. Ambos fármacos deberían tomarse con unos intervalos de separación de las comidas y entre sí de al menos una hora, lo que la hace muy poco práctica. Algunas publicaciones, sin embargo, sugieren buenos resultados de esta aproximación54,55b.
PosologíaD-penicilamina. Empezar con 250mg/día, aumentando cada semana 250mg hasta alcanzar una dosis de 1.000 (en las formas asintomáticas) o de 1.500mg/día (en los casos sintomáticos). Siempre las tomas deben estar separadas 1 o 2 h de las comidas, ya que la presencia de alimentos en el tubo intestinal impide la correcta absorción del fármaco. La administración cada 12 h favorece el cumplimiento del tratamiento. Cuando el paciente está asintomático se puede pasar a dosis de mantenimiento (500mg/12h). Conviene que los pacientes en tratamiento con penicilamina tomen piridoxina (25mg/día), ya que la penicilamina impide la disponibilidad de la piridoxina.
Trientina. Dosis de 750 a 1.500mg/día, en 2 tomas, separadas de las comidas, empezando con 250mg/día y también subiendo la dosis de semana en semana.
Sales de zinc. Dosis de 50mg/8 h, en los adultos, separadas de las comidas, al menos 1 h. Si el paciente nota molestias epigástricas, especialmente después de la dosis de la mañana, se puede recomendar que se haga la toma del medicamento con un poco de proteínas, como gelatina o un poco de jamón de York49. En los niños menores de 16 años se recomienda una dosis de 25mg/8 h.
¿Se puede reducir la dosis o el número de tomas?Se ha descrito una breve serie de 5 pacientes que por decisión propia tomaron 1.000mg/día de trientina en una sola toma y consiguieron la remisión de la enfermedad, sin efectos adversos56. La administración de una sola toma aumenta el cumplimiento del tratamiento, pero no existe ningún estudio que justifique esta pauta con la penicilamina. Con el cinc el tratamiento debe tomarse, al menos 50mg cada 12 h.
¿Cuáles son los efectos adversos posibles del tratamiento y cómo detectarlos?Los efectos secundarios son casi exclusivos de la penicilamina, y se registran en la tabla 2.
La trientina puede causar depresión medular, proteinuria y reacciones de carácter autoinmune pero de menor intensidad y con menor frecuencia que la penicilamina. Los efectos secundarios del cinc se limitan a molestias gástricas y sensación nauseosa.
Se ha descrito en pacientes tratados durante años, tanto con quelantes como con cinc, la aparición de un cuadro neurológico muy parecido a la polineuritis periférica por déficit de vitamina B12, relacionado con el déficit de cobre provocado por el tratamiento. Se manifiesta por una dificultad en la marcha, que se hace inestable, asociada a parestesias en manos y pies. El examen neurológico muestra hipoestesia en extremidades inferiores con hiperreflexia generalizada. Suele estar precedido por anemia y neutropenia, por lo que la observación de esta alteración hematológica obliga a investigar un posible déficit de cobre.
Si el cobre sérico no ligado a la ceruloplasmina es inferior a 5 μg/dl se debe sospechar esta situación que generalmente se acompaña de una cupruria de 24 h también muy baja. Los casos descritos de polineuropatía periférica sensitivo-motora habían sido tratados durante más de 13 años, con dosis generalmente elevadas de cinc57-62.
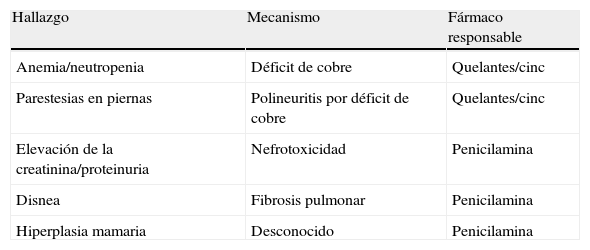
¿Cómo detectar los efectos adversos tardíos del tratamiento?Los efectos adversos a largo plazo son de difícil reconocimiento, ya que a veces son poco expresivos clínicamente o no se cae en la cuenta de que están relacionados con el tratamiento. El médico que atiende pacientes con EW debe sistemáticamente en las visitas periódicas revisar si aparece anemia, neutropenia, parestesias y pérdida de sensibilidad en piernas, o elevación de la creatinina, y sintomatología respiratoria (tos o disnea a grandes esfuerzos), que obligarían a descartar en el primer caso un eventual déficit de cobre y en el segundo toxicidad por penicilamina en forma de síndrome de Goodpasture o fibrosis pulmonar (tabla 3).
Signos de sospecha de la aparición de una complicación tardía relacionada con el tratamiento en la enfermedad de Wilson
| Hallazgo | Mecanismo | Fármaco responsable |
| Anemia/neutropenia | Déficit de cobre | Quelantes/cinc |
| Parestesias en piernas | Polineuritis por déficit de cobre | Quelantes/cinc |
| Elevación de la creatinina/proteinuria | Nefrotoxicidad | Penicilamina |
| Disnea | Fibrosis pulmonar | Penicilamina |
| Hiperplasia mamaria | Desconocido | Penicilamina |
Se debe sospechar fallo del tratamiento cuando no se consigue la mejoría de las manifestaciones clínicas o bioquímicas de la enfermedad. No obstante, antes de considerar un fallo de tratamiento debe el médico asegurarse de que el diagnóstico de EW era correcto. Si lo era debe, a continuación, excluirse un error en el cumplimiento del tratamiento o que la toma del fármaco era en dosis insuficiente.
Usualmente los pacientes precisan varios meses (de 4 a 6) para mostrar mejoría clínica o bioquímica (normalización de las transaminasas y reducción de la cupruria en los que la tenían elevada). En los pacientes con expresión neurológica la mejoría se puede demorar hasta los 2 años, pero después de este tiempo es improbable que se aprecie mayor mejoría. La disartria es el signo que experimenta menor tendencia a la regresión.
Durante el tratamiento debe monitorizarse periódicamente al paciente con determinación de transaminasas, ceruloplasmina, cupremia y cupruria para juzgar si el tratamiento es eficaz. El mejor criterio para asegurar que la enfermedad está compensada es la comprobación de que la cupremia libre está por debajo de 10 μg/dl. Una cupremia libre, es decir, el cobre no ligado a la ceruloplasmina, excesivamente baja (< 5 μg/dl), puede indicar una depleción de cobre causada por un excesivo efecto quelante.
¿Qué hacer si las transaminasas siguen altas en un paciente tratado?Las transaminasas suelen normalizarse entre 3 y 9 meses de iniciado el tratamiento, aunque persisten ligeramente altas en una proporción relativamente elevada de pacientes, en la mayoría de ellos por mal cumplimiento del mismo y en algunos por razones desconocidas en pacientes que hacen bien el tratamiento63.
En caso de encontrar las transaminasas altas debe seguirse la siguiente dinámica:
- a)
Comprobar que la enfermedad esté compensada, mediante la determinación del cobre libre en plasma. Si es inferior a 10 μg/dl debe considerarse que el paciente está bien tratado. Si no está bien compensado y el paciente estaba tratado con cinc se puede cambiar este fármaco por penicilamina63 o trientina68.
- b)
Si está bien tratado debe buscarse otra explicación a la elevación de las transaminasas, por ejemplo, otra enfermedad hepática, como hepatitis vírica crónica o enfermedad del hígado graso, concomitante33. En los pacientes tratados durante muchos años con penicilamina o con cinc puede haberse producido una depleción del cobre intrahepatocitario que induce una hemosiderosis, responsable de la hipertransaminasemia64. Este fenómeno es debido a una mayor reducción de la síntesis de ceruloplasmina causada por la deficiencia de cobre hepatocelular, que reduce a su vez la transformación del Fe++ a Fe+++ en el interior de los hepatocitos, indispensable para que el hierro se exporte de la célula hepática al plasma. Puede sospecharse por la elevación de la ferritina o mediante una RM abdominal, pero se confirma al comprobar una hemosiderosis en la biopsia hepática64-66. Este cuadro es similar al que se observa en la hipoceruloplasminemia congénita67.
Está justificado hacerlo en caso de intolerancia, por efectos secundarios, y en caso de ineficacia. La intolerancia al cinc, que se observa en algunos pacientes, es de tipo digestivo en forma de náuseas en la primera toma. La penicilamina puede causar disgeusia, que en algunos pacientes es motivo de solicitar cambiar de fármaco. La persistencia de la hipertransaminasemia, en ausencia de mal cumplimiento, indica ineficacia.
¿Se pueden tomar otros fármacos cuando se está tratando una enfermedad de Wilson?Sí, ya que no hay interferencia entre los fármacos utilizados en el tratamiento de la EW y los demás fármacos. Si el paciente toma trientina y hierro, las tomas de estos 2 medicamentos se deben separar al menos 2 h.
¿Cómo comprobar el cumplimiento?En los pacientes tratados con agentes quelantes se comprueba una excreción importante de cobre en la orina cuando toman el fármaco, en torno a 500 μg en 24h, aunque las fluctuaciones son notables en un mismo paciente. Niveles más bajos deben hacer sospechar mal cumplimiento o toma del fármaco demasiado próximo a las comidas, circunstancia que reduce la absorción de la penicilamina. En los tratados con cinc debe sospecharse el mal cumplimiento si persiste una excreción urinaria de cobre elevada. El examen de la concentración de cinc en la orina es también útil. Menos de 2mg/24 h indica mal cumplimiento.
No obstante, medir la concentración de cinc en sangre y orina puede ser engañoso, porque los valores altos podrían solo reflejar que el paciente se ha tomado el medicamento en los días previos al examen. Un estudio reciente muestra que el tratamiento asignado en cuanto a dosis y horario se incumple con mucha frecuencia y que el empeoramiento de la enfermedad solo se observa en pacientes que no cumplen adecuadamente69.
¿Se debe recomendar dieta?Algunos alimentos, como el chocolate, el hígado, los mariscos y las setas, contienen elevadas concentraciones de cobre, por lo que es mejor abstenerse de comerlos, pero es improbable que un consumo ocasional comporte algún perjuicio, especialmente en los pacientes tratados con cinc, ya que este impedirá la absorción intestinal del cobre contenido en estos alimentos.
Algunos estudios experimentales indican que las proteínas de soja pueden estimular el daño hepático de las ratas con un modelo de EW70. Aunque no existe ninguna demostración de este efecto en humanos no sería ninguna irracionalidad recomendar restringir el consumo de soja en los pacientes con EW.
¿Se debe hacer una biopsia hepática durante el seguimiento de un paciente tratado?No tiene sentido hacerla, ya que disponemos de otros medios para reconocer el mal cumplimiento en la toma de la medicación y la eficacia del tratamiento. Por otra parte, en la mayoría de pacientes bien tratados no se reduce en exceso la concentración de cobre en el tejido hepático, ya que los fármacos que utilizamos, penicilamina, trientina y cinc, ejercen su efecto beneficioso mayoritariamente por la estimulación de la síntesis de metalotioneína en los tejidos, que fija el cobre en posición atóxica. Solo al cabo de muchos años de tratamiento con cinc se puede observar una disminución del cobre intrahepático62.
¿Cuándo estaría indicado el trasplante hepático?El trasplante está indicado en los pacientes que presentan un cuadro de insuficiencia hepática aguda grave, en los pacientes cuya enfermedad se manifiesta con alguna complicación de una cirrosis hepática o cuando la enfermedad hepática es progresiva y se descompensa a pesar del tratamiento o por el mal cumplimiento por parte del paciente con la medicación71. No está justificado como tratamiento del síndrome neurológico de la enfermedad72, aunque en algunos pacientes con EW neurológica el trasplante hepático permitió la regresión de las manifestaciones clínicas73,74.
El trasplante es efectivo en la mayoría de pacientes y permite no tomar más la medicación quelante o el cinc. En los casos trasplantados por insuficiencia hepática grave la supervivencia es del orden del 75% y en los casos de enfermedad hepática avanzada de cerca del 90%. El trasplante de donante vivo da buen resultado a pesar de que muchos de ellos sean heterocigotos para el defecto de la enfermedad75.
Otras preguntas frecuentes¿Qué frecuencia de visitas hay que recomendar en el seguimiento y qué tipo de exámenes?Al inicio del tratamiento con penicilamina, se le deben explicar al paciente los signos de una posible reacción alérgica o la posibilidad de aparición de alteraciones neurológicas, para que contacten de forma inmediata con el médico. Si no ocurren signos de toxicidad inicial es recomendable establecer una visita de control en 2-3 meses.
Posteriormente, los pacientes con EW deben ser visitados con una relativa frecuencia (por ejemplo, cada 6 meses) con los siguientes objetivos:
- a)
Comprobar si el tratamiento prescrito es eficaz y bien tolerado (valoración clínica y medición de los niveles de cobre libre en sangre).
- b)
Comprobar el cumplimiento con la medición de cobre y cinc urinarios.
- c)
Asegurar que comprenden la naturaleza de su enfermedad y la necesidad de seguir tratamiento toda la vida.
- d)
Hacer prevención de la depresión que afecta a algunos pacientes al conocer la enfermedad que presentan y que en ocasiones puede inducir al suicidio76.
Durante el embarazo no debe interrumpirse la medicación. Podría causar la aparición de un fallo hepático fulminante77. Ni los quelantes ni las sales de cinc causan efectos adversos en la madre y el feto78. Si la paciente toma penicilamina puede ser útil reducir a 500mg la dosis de penicilamina para facilitar una adecuada cicatrización de la herida en caso de precisar cesárea79. La trientina no ejerce ningún efecto negativo80.
¿Cuál es el mejor método de anticoncepción?La mayoría de pacientes con EW son fértiles y pueden quedar embarazadas. Cuando precisan consejo acerca de cómo evitar el embarazo se puede recomendar espermicidas y métodos de barrera, y preparados de progesterona81, pues no tienen ninguna influencia negativa en el metabolismo del cobre. Los estrógenos pueden reducir la excreción biliar82 y, por tanto, la eliminación del cobre, y los dispositivos intrauterinos pueden contener cobre.
¿De qué fallecen los pacientes con enfermedad de Wilson?Con el tratamiento adecuado los pacientes con EW tienen la expectativa de vida igual que las personas de su edad. Los pacientes diagnosticados en la fase de enfermedad hepática avanzada, como cirrosis, pueden fallecer por alguna de las complicaciones de la enfermedad antes si no se efectúa un trasplante hepático. Otros pacientes fallecen prematuramente por fallo diagnóstico, finalmente los pacientes que no cumplen o abandonan el tratamiento están expuestos a experimentar complicaciones y muerte relacionada con la enfermedad83.
¿A qué familiares de un paciente recién diagnosticado de enfermedad de Wilson hay de investigar y cómo?Hay que examinar todos sus hermanos y también a sus padres si estos presentan alguna manifestación de enfermedad hepática o neurológica, que pudiera ser una EW. Lo ideal sería buscar si existe la misma mutación que en el caso índice, sin necesidad de secuenciar todos los exones del gen, ya que los miembros afectados de una familia presentan el mismo fenotipo.
Si no se dispone de estudio genético el reconocimiento de casos presintomáticos puede hacerse mediante la determinación de las transaminasas, la ceruloplasmina y la cupruria de 24h. Si estas pruebas son normales el diagnóstico de EW es improbable, aunque para mayor seguridad puede ser útil recomprobar al cabo de unos años que siguen siendo normales.
El examen de los hermanos del caso índice no debe hacerse en niños menores de 3 años, ya que la enfermedad no suele presentarse antes de esta edad.
La trientina no está comercializada en España. Se puede conseguir como medicamento de uso compasivo.

