







Las secuencias de proteasa y transcriptasa reversa del VIH-1 aportan una información muy valiosa para el manejo de la infección por VIH, más allá de la información de resistencias a los antirretrovirales. En nuestro estudio la hemos utilizado para evaluar las cadenas de transmisión, la transmisión de resistencias entre ellos, y para conocer la distribución espacial de los diferentes subtipos utilizando técnicas de georreferenciación.
MétodosHemos estudiado 693 pacientes diagnosticados de VIH-1 durante el periodo 2005-2012, todos ellos residentes en Andalucía Oriental. La secuencia del gen pol (transcriptasa reversa y proteasa) se generó utilizando Trugene® HIV Genotyping Kit (Siemens, NAD). La historia evolutiva fue inferida a través de MEGA 5.2 mediante el método de Neighbor-Joining. Para la filogeografía y el estudio de resistencias utilizamos ArcGIS y REGA.
ResultadosDoscientos noventa y ocho pacientes se asociaron en 77 clusters diferentes. La mayoría de los cluster estaban formados por parejas (n=49), de hombres que practican sexo con hombres (n=26), de nacionalidad española (n=37), con una edad menor a 45 años (73,5%). Las áreas de mayor heterogeneidad de subtipos fueron el área metropolitana de Granada y las zonas de costa de Almería y Granada. Hemos encontrado 5 cluster con más de 10 individuos. En 15 cluster detectamos mutaciones de resistencia.
ConclusionesPresentamos datos que demuestran que el estudio epidemiológico de los diferentes clusters de transmisión de VIH mediante análisis filogenético se presenta como una herramienta potente y de gran utilidad para la vigilancia y control epidemiológico de la propagación del VIH, que puede ayudar a diseñar actuaciones eficaces para prevenir la diseminación del VIH.
Protease and reverse transcriptase HIV-1 sequences provide useful information for patient clinical management, as well as information on resistance to antiretrovirals. The aim of this study is to evaluate transmission events, transmitted drug resistance, and to georeference subtypes among newly diagnosed patients referred to our center.
MethodsA study was conducted on 693 patients diagnosed between 2005 and 2012 in Southern Spain. Protease and reverse transcriptase sequences were obtained for resistance to cART analysis with Trugene® HIV Genotyping Kit (Siemens, NAD). MEGA 5.2, Neighbor-Joining, ArcGIS and REGA were used for subsequent analysis.
ResultsThe results showed 298 patients clustered into 77 different transmission events. Most of the clusters were formed by pairs (n=49), of men having sex with men (n=26), Spanish (n=37), and below 45 years of age (73.5%). Urban areas from Granada, and the coastal areas of Almeria and Granada showed the greatest subtype heterogeneity. Five clusters were formed by more than 10 patients, and 15 clusters had transmitted drug resistance.
ConclusionsThe study data demonstrate how the phylogenetic characterization of transmission clusters is a powerful tool to monitor the spread of HIV, and may contribute to design correct preventive measures to minimize it.
La infección por el virus de la inmunodeficiencia humana (VIH) ha pasado de ser una pandemia controlada a experimentar un incremento en el número de casos en países con un nivel económico y sanitario adecuado, especialmente en determinados colectivos, como en el de hombres que tienen sexo con hombres (HSH)1–5. Los cambios de comportamiento, consecuencia de vivir en una era de terapia antiviral eficiente, han contribuido al incremento de la propagación del VIH. El resultado de estudiar la asociación de la diversidad viral con diferentes parámetros demográficos evidencia una epidemia muy compartimentada, determinada por las características sociales y los hábitos de comportamiento de estos pacientes6.
La epidemiología molecular, a través del estudio de las características de clusters bien definidos7–10, puede ayudar a controlar estas subepidemias. El uso de técnicas de análisis filogenético en grandes estudios de cohorte para el control de la propagación del VIH se plantea como una potente herramienta de alerta epidemiológica, tanto para la contención de cadenas de transmisión de VIH, en donde el virus se puede estar transmitiendo a una alta velocidad9,10, como para el control de la propagación de mutaciones de resistencia (TDR) dentro de grupos donde existe un especial riesgo de transmisión. Además, otras herramientas utilizadas en la filogeografía, como la georreferenciación, permiten situar espacialmente los diversos integrantes que conforman las diferentes cadenas de transmisión y así poder analizar las zonas geográficas donde existe una mayor recombinación para el VIH.
Cada vez es mayor el número de investigaciones que utilizan las técnicas de análisis filogenético en estudios de cohorte, analizando la situación local, con el objetivo de poder establecer las adecuadas medidas de contención11–14. Sin embargo, en España existen pocos estudios que aborden este problema mediante métodos de epidemiología molecular. Nuestro centro sirve de referencia para el estudio de resistencia a antirretrovirales a toda el área de Andalucía Oriental (Granada, Almería y Jaén). Disponemos de información de la secuencia del gen pol (transcriptasa reversa y proteasa), información que se encuentra ligada a datos clínicos a través de un grupo colaborativo con las unidades de enfermedades infecciosas. Por ello, en este escenario nos hemos planteado describir el panorama actual del VIH en Andalucía Oriental mediante el estudio epidemiológico pormenorizado de las diferentes cadenas de transmisión de VIH, prestando especial interés a aquellas con resistencias a los fármacos antirretrovirales. Como objetivo secundario hemos analizado la heterogeneidad existente entre los diferentes subtipos que integran los clusters, a través de la representación espacial de estos subtipos mediante técnicas de georreferenciación.
Pacientes y métodosEl estudio ha incluido a 693 pacientes naïve, nuevos diagnósticos de VIH de los hospitales de Granada, Almería y Jaén, durante el periodo 2005-2012. De estos pacientes disponíamos de una secuencia fasta del gen pol (posiciones 4-99 de la proteasa y 38-247 de la transcriptasa reversa), obtenidas utilizando el ensayo Trugene® HIV-1 Genotyping Kit (Siemens [NAD]).
Estas secuencias fueron procesadas y alineadas utilizando MEGA 5.215, utilizando el método de Neighbor-Joining para inferir la historia evolutiva. Para su representación, el árbol filogenético bootstrap (medida de soporte y fiabilidad para los diferentes nodos de cada uno de los clusters) de consenso fue inferido a partir de 1.000 réplicas. Las ramas pertenecientes a los clusters con un valor de bootstrap inferior al 80% de las réplicas fueron eliminadas. Las distancias evolutivas se calcularon utilizando el método de Kimura 2 parámetros. La tasa de variación entre sitios fue modelada con una distribución gamma equivalente a 2,1, utilizando FindModel para modelar la evolución nucleotídica. Para la construcción de un modelo resumido del árbol filogenético generado en MEGA 5.2 utilizamos FigTree v. 1.4.0 (disponible en: http://tree.bio.ed.ac.uk/software/figtree).
Se realizó un estudio epidemiológico pormenorizado de los clusters con un valor de bootstrap superior al 85%, analizando los siguientes parámetros demográficos, clínicos y virológicos: fecha de nacimiento, año de diagnóstico de VIH, hospital de referencia, sexo, mecanismo de transmisión de VIH, subtipo, país de nacimiento y lugar de residencia actual.
Para el análisis de las resistencias asociadas a los clusters hemos utilizado las mutaciones de la lista de la OMS16 y el algoritmo de interpretación de resistencias de la Universidad de Stanford17. La determinación del subtipo viral se realizó utilizando REGA HIV-1 Subtyping Tool v. 2.018. Para la georreferenciación de los diferentes subtipos VIH de los pacientes incluidos en todos los clusters utilizamos ArcGIS Explorer Desktop Esri®, un visor con sistema de información geográfica, que nos permitió realizar un análisis espacial, de proximidad y la edición de un mapa personalizado, en nuestro caso, para el estudio de zonas geográficas con una mayor prevalencia y heterogeneidad de subtipos.
ResultadosLos pacientes estudiados fueron mayoritariamente hombres jóvenes (41 años), HSH (51,5%), nativos españoles (65,5%), que residen en la zona de Granada (43,7%) o Almería (36,9%) y que están infectados por subtipos B (65,2%).
De las 693 secuencias utilizadas para generar el árbol filogenético, 298 (43%) se agruparon en 77 clusters diferentes, con un valor medio de bootstrap de 97,12%. La gran mayoría de estos estaban formados por pares (63,7%), fundamentalmente por pares de HSH (53,1%).
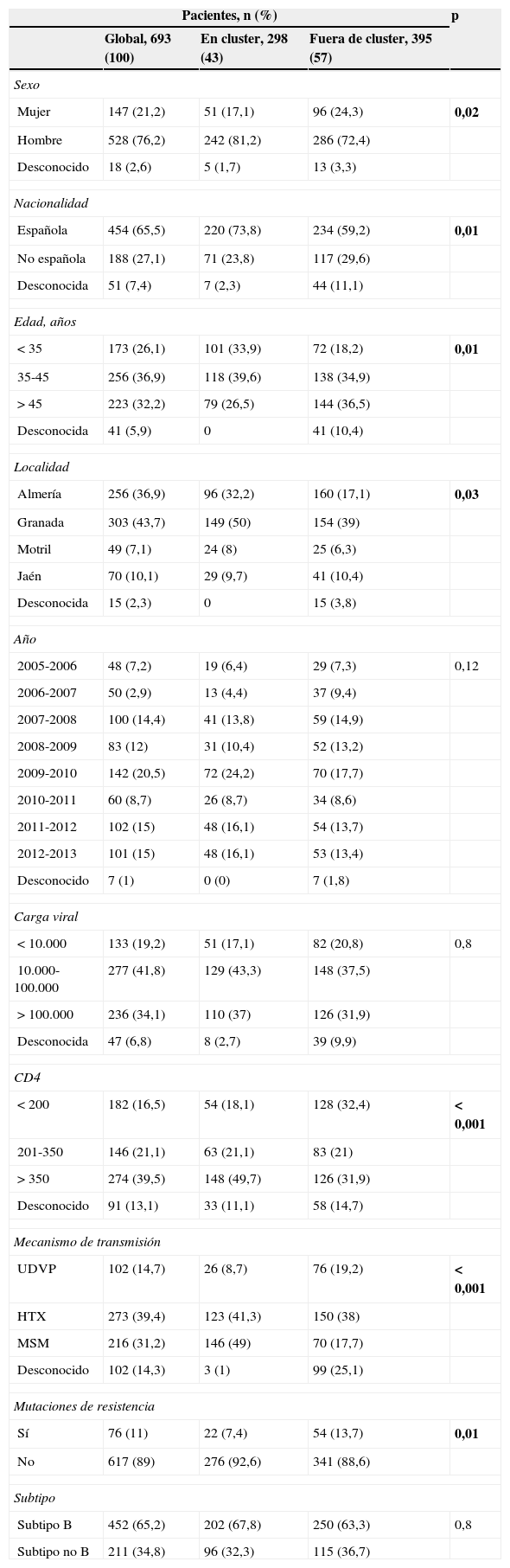
Las características clínicas, virológicas y demográficas de los pacientes se muestran en la tabla 1. Existieron diferencias estadísticamente significativas entre los pacientes asociados en cluster y los que no lo estaban. En concreto, la asociación en cluster fue más frecuente en hombres (p=0,02), españoles (p=0,01), con una edad menor de 35 años (p=0,01), residentes en Granada (p=0,03), con un número de CD4 mayor de 350 (p<0,001), homosexuales (p<0,001) y que no presentaban mutaciones de resistencia a antirretrovirales de primera línea (p=0,01).
Características clínicas, demográficas y virológicas de los pacientes incluidos en cluster vs. pacientes fuera de cluster
| Pacientes, n (%) | p | |||
|---|---|---|---|---|
| Global, 693 (100) | En cluster, 298 (43) | Fuera de cluster, 395 (57) | ||
| Sexo | ||||
| Mujer | 147 (21,2) | 51 (17,1) | 96 (24,3) | 0,02 |
| Hombre | 528 (76,2) | 242 (81,2) | 286 (72,4) | |
| Desconocido | 18 (2,6) | 5 (1,7) | 13 (3,3) | |
| Nacionalidad | ||||
| Española | 454 (65,5) | 220 (73,8) | 234 (59,2) | 0,01 |
| No española | 188 (27,1) | 71 (23,8) | 117 (29,6) | |
| Desconocida | 51 (7,4) | 7 (2,3) | 44 (11,1) | |
| Edad, años | ||||
| <35 | 173 (26,1) | 101 (33,9) | 72 (18,2) | 0,01 |
| 35-45 | 256 (36,9) | 118 (39,6) | 138 (34,9) | |
| >45 | 223 (32,2) | 79 (26,5) | 144 (36,5) | |
| Desconocida | 41 (5,9) | 0 | 41 (10,4) | |
| Localidad | ||||
| Almería | 256 (36,9) | 96 (32,2) | 160 (17,1) | 0,03 |
| Granada | 303 (43,7) | 149 (50) | 154 (39) | |
| Motril | 49 (7,1) | 24 (8) | 25 (6,3) | |
| Jaén | 70 (10,1) | 29 (9,7) | 41 (10,4) | |
| Desconocida | 15 (2,3) | 0 | 15 (3,8) | |
| Año | ||||
| 2005-2006 | 48 (7,2) | 19 (6,4) | 29 (7,3) | 0,12 |
| 2006-2007 | 50 (2,9) | 13 (4,4) | 37 (9,4) | |
| 2007-2008 | 100 (14,4) | 41 (13,8) | 59 (14,9) | |
| 2008-2009 | 83 (12) | 31 (10,4) | 52 (13,2) | |
| 2009-2010 | 142 (20,5) | 72 (24,2) | 70 (17,7) | |
| 2010-2011 | 60 (8,7) | 26 (8,7) | 34 (8,6) | |
| 2011-2012 | 102 (15) | 48 (16,1) | 54 (13,7) | |
| 2012-2013 | 101 (15) | 48 (16,1) | 53 (13,4) | |
| Desconocido | 7 (1) | 0 (0) | 7 (1,8) | |
| Carga viral | ||||
| <10.000 | 133 (19,2) | 51 (17,1) | 82 (20,8) | 0,8 |
| 10.000-100.000 | 277 (41,8) | 129 (43,3) | 148 (37,5) | |
| >100.000 | 236 (34,1) | 110 (37) | 126 (31,9) | |
| Desconocida | 47 (6,8) | 8 (2,7) | 39 (9,9) | |
| CD4 | ||||
| <200 | 182 (16,5) | 54 (18,1) | 128 (32,4) | <0,001 |
| 201-350 | 146 (21,1) | 63 (21,1) | 83 (21) | |
| >350 | 274 (39,5) | 148 (49,7) | 126 (31,9) | |
| Desconocido | 91 (13,1) | 33 (11,1) | 58 (14,7) | |
| Mecanismo de transmisión | ||||
| UDVP | 102 (14,7) | 26 (8,7) | 76 (19,2) | <0,001 |
| HTX | 273 (39,4) | 123 (41,3) | 150 (38) | |
| MSM | 216 (31,2) | 146 (49) | 70 (17,7) | |
| Desconocido | 102 (14,3) | 3 (1) | 99 (25,1) | |
| Mutaciones de resistencia | ||||
| Sí | 76 (11) | 22 (7,4) | 54 (13,7) | 0,01 |
| No | 617 (89) | 276 (92,6) | 341 (88,6) | |
| Subtipo | ||||
| Subtipo B | 452 (65,2) | 202 (67,8) | 250 (63,3) | 0,8 |
| Subtipo no B | 211 (34,8) | 96 (32,3) | 115 (36,7) | |
En negrita, asociaciones con significación estadística.
El 48% de los clusters estaba formado por nativos españoles, el 23% incluía algún extranjero entre sus integrantes y solo el 3% estaba compuesto íntegramente por extranjeros. Se localizaban más frecuentemente entre pacientes con menos de 45 años (73,5%), que viven en el área metropolitana de Granada (31,6%). En Andalucía Oriental los episodios de transmisión se producen más frecuentemente entre pacientes infectados con subtipos B (66%), y el 22% se producen entre subtipos no B. El 12% restante se produjo entre pacientes infectados con diferente subtipo (subtipo B y recombinante B-D).
Hemos detectado transmisión de mutaciones de resistencia para fármacos antirretrovirales de primera línea en 15 clusters diferentes, generalmente en los de tamaño pequeño. Para los análogos de nucleósidos se detectaron mutaciones en 3 clusters diferentes frente a análogos de timidina (TAMS, M41L; D67N+K219Q; K70R, respectivamente), en 2 cambios en la posición M184 (M184I, en uno; M184V, en otro), en un cluster se detectó la revertiente T215L, y en otro, la combinación D67E+F77L. Las mutaciones para los NNRTI fueron las más frecuentes, detectándose en un total de 7 clusters; su distribución fue la siguiente: K101E; V179D; P225H; V90I+Y181C+H221Y; K103N; V179E; E138A+V179D. Finalmente, 5 clusters mostraron mutaciones de resistencia en la proteasa: M46I (3 clusters) y L90M (2 clusters), y 2 clusters mostraron mutaciones de resistencia frente a más de una familia de antirretrovirales.
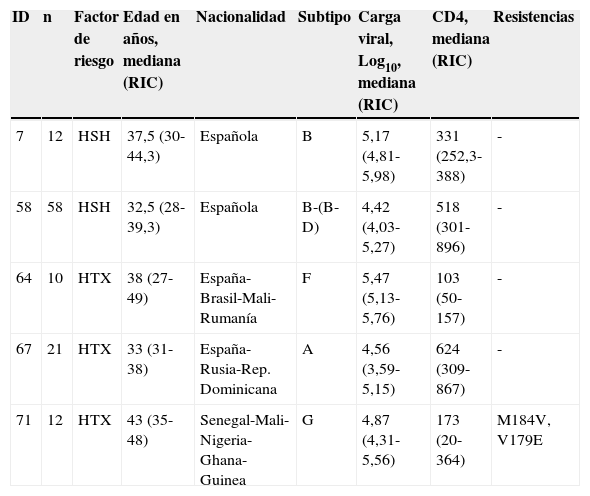
Hemos detectado 5 cadenas de transmisión en las que estaba incluido un número igual o mayor a 10 pacientes, que representaban un 16% de los individuos de nuestra cohorte y estaban integrados por 113 individuos. La tabla 2 muestra las principales características de cada uno de estos clusters, y las figuras 1 y 2, el árbol filogenético de la población estudiada. En Andalucía, los clusters con mayor repercusión epidemiológica y que involucran extranjeros entre sus integrantes (Europa del Este, América Latina, África) son todos de población HTX y con subtipos no B; en cambio, los que involucran individuos españoles son todos de población HSH y con subtipo B. Cabe destacar, por su número, una cadena de transmisión formada por 58 HSH de nacionalidad española que se inicia en el año 2007 e incorpora individuos hasta el año 2012, y otro de 21 pacientes HTX formado por varones de nacionalidad española y mujeres de nacionalidad rusa, localizado en la provincia de Almería. Asimismo, destaca también un cluster formado por 12 pacientes de Senegal, Mali, Nigeria, Ghana y Guinea, en el que se detecta la transmisión de M184V y V179D, con resistencia a los análogos de primera línea emtricitabina y lamivudina, y resistencia de bajo nivel a abacavir.
Características clínicas, demográficas y virológicas de los clusters de transmisión con mayor relevancia epidemiológica
| ID | n | Factor de riesgo | Edad en años, mediana (RIC) | Nacionalidad | Subtipo | Carga viral, Log10, mediana (RIC) | CD4, mediana (RIC) | Resistencias |
|---|---|---|---|---|---|---|---|---|
| 7 | 12 | HSH | 37,5 (30-44,3) | Española | B | 5,17 (4,81-5,98) | 331 (252,3-388) | - |
| 58 | 58 | HSH | 32,5 (28-39,3) | Española | B-(B-D) | 4,42 (4,03-5,27) | 518 (301-896) | - |
| 64 | 10 | HTX | 38 (27-49) | España-Brasil-Mali-Rumanía | F | 5,47 (5,13-5,76) | 103 (50-157) | - |
| 67 | 21 | HTX | 33 (31-38) | España-Rusia-Rep. Dominicana | A | 4,56 (3,59-5,15) | 624 (309-867) | - |
| 71 | 12 | HTX | 43 (35-48) | Senegal-Mali-Nigeria-Ghana-Guinea | G | 4,87 (4,31-5,56) | 173 (20-364) | M184V, V179E |

 de los diferentes clusters de transmisión del árbol filogenético consenso.")
Las áreas de mayor prevalencia de VIH por km2 se correspondían, a su vez, con las zonas en las que existía una mayor heterogeneidad de subtipos, destacando la zona metropolitana de Granada y las zonas de costa de Almería y Granada. Los individuos que estaban localizados en estas áreas mostraban, a su vez, una mayor agregación, basándonos en la distancia entre individuos que nos facilitaba el visor con sistema de información geográfica de ArcGIS. Estos datos se representan gráficamente en la figura 3.
Discusión
En Andalucía Oriental, la asociación en clusters entre pacientes con infección por VIH ocurre con mayor frecuencia en pares de HSH, españoles, residentes en Granada, con menos de 35 años, y con menor prevalencia de transmisión de mutaciones de resistencia.
Los datos aportados en este estudio son similares a los descritos en otras series8, que también comunican una clara tendencia a la agrupación de los clusters entre pares de HSH. El hallazgo de grandes cadenas de transmisión, encontradas con mayor frecuencia en HSH jóvenes, evidencian en determinados colectivos una rápida y eficaz propagación del VIH. Yebra et al.7 demuestran cómo en la Comunidad de Madrid se produce una mayor inclusión de HSH en estas cadenas de transmisión. Leigh Brown et al.9 destacan que hasta un 29% de la población en Reino Unido se agruparía en grandes cadenas de transmisión con más de 10 individuos, y Ambrosioni et al.8 aportan datos de la cohorte suiza de VIH, en la cual, un 42% de los individuos pertenecen a cadenas de transmisión, por lo que reivindican que los HSH con infección reciente por VIH son una fuente importante de transmisión y que las campañas de prevención deberían centrarse en mejorar el diagnóstico de los individuos con infección reciente.
El mayor riesgo de transmisión de VIH en Andalucía Oriental se produce entre pacientes que oscilan entre los 35 y los 50 años, datos que concuerdan con los reflejados en el Centro de Vigilancia Epidemiológico de VIH de España para otras comunidades autónomas19. Asimismo, en consonancia con el aumento de la población inmigrante que ocurrió en los últimos años en España, hemos visto cómo en Andalucía Oriental hasta el 23% de las cadenas de transmisión están formadas por españoles y no españoles conjuntamente; además, las cadenas de transmisión con mayor repercusión epidemiológica en la población HTX estaban integradas por una gran proporción de individuos extranjeros, como es el caso del cluster 67, formado por 21 individuos de subtipo A, y por un gran porcentaje de mujeres de nacionalidad rusa. Este hecho subraya la necesidad de diversificar los programas de prevención para adaptarlos a las necesidades de este colectivo, social y culturalmente muy heterogéneo, y especialmente vulnerable. Aunque se han encontrado episodios de transmisión de resistencias a antirretrovirales de primera línea, como en otros estudios, en la mayoría de las ocasiones se detectan entre pares («diadas»)20,21, y solo en un caso se han agregado en una cadena de transmisión que incluía a 12 pacientes. La vigilancia de los episodios de transmisión y la contribución a la epidemiología molecular por parte de los centros asistenciales en los que se determinan resistencias a los antirretrovirales son fundamentales para alertar y generar esfuerzos colaborativos con los servicios de Medicina Preventiva y Enfermedades Infecciosas, y con las autoridades sanitarias, para elevar las medidas de contención en la propagación de la transmisión de resistencias.
Nuestro estudio presenta algunas limitaciones: en primer lugar, como es frecuente en estudios retrospectivos, no hemos podido recopilar todos los datos demográficos en los pacientes que hemos estudiado; de especial importancia ha sido no poder disponer de la proporción de individuos con infección aguda/reciente, ya que los niveles de carga viral y de CD4 de los clusters mayoritarios de HSH con subtipo B evidencian que probablemente existe un mayor número de infecciones recientes entre estos pacientes. En segundo lugar, en lo referente al estudio de las resistencias, hemos basado nuestras determinaciones en secuenciación de poblaciones, no habiendo investigado aquellas resistencias que pudieran estar presentes en formas minoritarias. Por último, hemos elegido un valor de bootstrap del 85% para definir los clusters de transmisión, punto de corte que puede parecer bajo; aun así, la mayoría de los pacientes estaban incluidos en clusters de transmisión con valores de bootstrap cercanos al 100%, y bajamos el punto de corte para evitar perder individuos pertenecientes a cadenas de transmisión con un tamaño muy grande, y que presentaban evidencias clínicas y demográficas de estar incluidos en ellas. En general, valores de bootstrap por encima del 70%22 para cadenas de transmisión grandes serían fiables, a pesar de lo cual, asumimos el hecho de que unos pocos individuos pudieran quedar fuera de alguna cadena de transmisión.
En resumen, en nuestro trabajo presentamos datos que demuestran que el estudio epidemiológico de las diferentes cadenas de transmisión de VIH mediante análisis filogenético se presenta como una herramienta potente y de gran utilizad para la vigilancia y control epidemiológico de la propagación del VIH, y que su utilización ayudaría a reforzar e implantar actuaciones eficaces para prevenir la diseminación del VIH.
FinanciaciónEste trabajo ha sido financiado por el proyecto RD12/0017/006 integrado en el Plan Nacional de I+D+i y cofinanciado por el ISCIII-Subdirección General de Evaluación y el Fondo Europeo de Desarrollo Regional (FEDER), y por el PI12/01053. También ha sido financiado en parte por la Consejería de Igualdad, Salud y Políticas Sociales, Junta de Andalucía (AC-0082-2013).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

