







The MC4R gene plays a critical role in regulating food intake, making it an important model for studying genetic mutations that impact the protein function. This study aimed to identify the most deleterious functional and structural variants in individuals with obesity by analyzing SNPs from the NCBI dbSNP database and selecting pathogenic variants from ClinVar. Bioinformatics tools were employed to predict deleterious SNPs, and conservation analysis was performed using ConSurf. Stability predictions were made with MUpro, I-Mutant2.0, and iStable. The 3D structure of the MC4R protein was examined using YASARA view. A total of 20 out of 348 missense mutations were associated with obesity. Fifteen of these variants were predicted to be the most deleterious. Eight variants located in conserved regions were found to significantly reduce protein stability and cause structural changes (S58C, E61K, N62S, I69R, D90N, R165Q, P299H, and I316S), indicating their potential as obesity biomarkers and therapeutic targets.
El gen MC4R desempeña un papel crítico en la regulación de la ingesta de alimentos, por lo que es un modelo importante para estudiar las mutaciones genéticas que afectan a la función de la proteína. Este estudio tenía como objetivo identificar las variantes funcionales y estructurales más deletéreas en individuos con obesidad mediante el análisis de polimorfismos de la base de datos NCBI dbSNP y la selección de variantes patogénicas de ClinVar. Se emplearon herramientas bioinformáticas para predecir SNPs deletéreos, y el análisis de conservación se realizó con ConSurf. Las predicciones de estabilidad se realizaron con MUpro, I-Mutant2.0 e iStable. La estructura 3D de la proteína MC4R se examinó con YASARA view. Un total de 20 de las 348 mutaciones sin sentido se asociaron con la obesidad. Se predijo que 15 de estas variantes eran las más deletéreas. Se observó que ocho variantes localizadas en regiones conservadas reducían significativamente la estabilidad de la proteína y provocaban cambios estructurales (S58C, E61K, N62S, I69R, D90N, R165Q, P299H e I316S), lo que indica su potencial como biomarcadores de obesidad y dianas terapéuticas.
Monogenic forms of obesity are mainly induced by genetic mutations in the coding sequence of the melanocortin-4 receptor (MC4R), resulting in a deficiency of this gene and autosomal co-dominant inheritance with incomplete penetrance.1 Indeed, Stutzmann et al. (2008) showed that penetrance varies with age: 79% in children, 60% in adults aged 18–52, and 40% in adults older than 52 years.2 Furthermore, Farooqi et al. (2003) found that a more obesogenic environment exacerbates the impact of the MC4R mutation, leading to a more severe phenotype and higher loss of function, and vice versa.3 These mutations are responsible for up to 6% of individuals with early-onset severe obesity.4
The MC4R gene is situated at the 18q21.32 position on the chromosome and contains a single exon that encodes a protein of 332 amino acids. This protein belongs to the G protein-coupled receptor (GPCR) family. The MC4R protein is essential for regulating energy homeostasis, food intake, and body weight by interacting with its ligand, alpha-melanocyte stimulating hormone (α-MSH).5 Activation of the MC4R receptor leads to a conformational change. This active conformation induces the interaction of the receptor with various G proteins, particularly the Gs (stimuli) and Gq proteins, which are involved in the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) and calcium pathways, respectively. α-MSH increases energy expenditure by regulating intracellular cAMP concentrations through increasing adenylate cyclase (AC) activity.6 The resulting cAMP accumulation activates PKA, followed by Exchange protein directly activated by cAMP (EPAC), ERK1/2, and cAMP response element-binding protein (CREB), leading to increased c-Fos transcription and decreased phosphorylation and AMPK activity. Gs signaling occurs in the dorsomedial nucleus to regulate energy expenditure.4,7,8
Ligand binding to its receptor also induces phospholipase C (PLC) activation and an increase in cytosolic Ca2+ concentration via Gq, resulting in a decrease in food intake. This Gq signaling takes place in the paraventricular nucleus of the hypothalamus to control food intake.9 α-MSH signaling also increases the excitability of MC4R neurons by regulating potassium channel activity.10 Ligand binding induces Kir7.1 closure, causing depolarization of MC4R neurons and regulating energy homeostasis.11 The agouti-related protein (AgRP)—a competitive antagonist of α-MSH—inhibits AC pathway activation by Gs. In addition, AgRP can act as an agonist to promote MC4R signaling by Gi (inhibition), which plays an inhibitory role in this pathway.8 Furthermore, AgRP promotes the hyperpolarization of MC4R neurons by opening the Kir7.1 channel, thereby decreasing the activity of the receptor.9,12
To date, over 200 mutations have been identified in populations from the Maghreb, Europe, America, and Asia.11,13–26 Mutations leading to either complete or partial loss of function in the MC4R gene can disrupt cAMP production, thus contributing to the most common monogenic form of obesity.27,28 However, around 25% of MC4R mutations have been categorized as wild-type or non-pathogenic, as they do not reduce cAMP production.8 Individuals carrying homozygous or compound heterozygous mutations are rare, with only a few cases being reported from consanguineous families.1,3,29–32 These individuals exhibited morbid obesity vs their heterozygous parents who had intermediate phenotypes.1
Therefore, this study aimed to evaluate previously identified mutations in individuals with obesity to identify the most functionally and structurally deleterious variants.
MethodsData set collectionThe NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/SNP/) was used to access the MC4R gene SNPs. Only missense variants were selected from dbSNP. Subsequently, SNPs presenting obesity as the main clinical feature with pathogenic clinical significance were selected from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). ClinVar is a database of genetic variants that are interpreted according to their clinical significance.
Prediction of deleterious SNPs by bioinformatics toolsTo predict the possible impact of MC4R gene variants selected on NCBI on protein function, we used various bioinformatics tools available online. These algorithmic programs include Polymorphism Phenotyping Version 2 (PolyPhen-2: www.genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT: www.sift.bii.a-star.edu.sg/), SNAP2 (www.rostlab.org/services/snap2web/), PON-P2 (www.structure.bmc.lu.se/PON-P2/), Meta-SNP (www.snps.biofold.org/meta-snp/), Predicting disease-associated variations using GO terms (SNPs&GO: https://snps.biofold.org/snps-and-go/snps-and-go.html) and PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp1/).
The protein sequence (P32245) encoded by the MC4R gene and used for bioinformatics software was obtained from the UniProt database (https://www.uniprot.org/).
Conservation analysisConservation analysis was conducted via the ConSurf web server (http://consurf.tau.ac.il/). This widely used tool reveals the functional regions of proteins by analyzing the evolutionary dynamics of amino acid substitutions among homologous sequences. ConSurf estimates amino acid evolution rates.33
Prediction of SNPs effect on protein stabilityMUpro, I-Mutant2.0, and iStable are computational tools designed to predict the effects of mutations on protein stability. Each tool uses different algorithms and approaches to assess how a single-point mutation might affect the stability of a protein structure. MUpro (http://mupro.proteomics.ics.uci.edu/) uses Support Vector Machines (SVMs) and Neural Networks. I-Mutant2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html) is based on Support Vector Machines (SVMs). The stability depends on the free energy change (Delta Delta G: ΔΔG (kcal/mol)) based on the protein sequence. Scores>0 indicate a decrease in stability, while scores>0 indicate an increase. ΔΔG values can be categorized into 3 predictions: largely unstable (ΔΔG<−0.5kcal/mol), largely stable (ΔΔG>0.5kcal/mol), or neutral (−0.5≤ΔΔG≤0.5kcal/mol).34,35 iStable (http://ncblab.nchu.edu.tw/iStable2) combines predictions from multiple machine learning-based tools, including MUpro and I-Mutant2.0, among others. It provides a confidence score between 0 and 1, with higher scores being indicative of greater confidence in the prediction.36
HOPEThe web service tool Have (y)Our Protein Explained (HOPE) (https://www3.cmbi.umcn.nl/hope/) examines the structural effects of a point mutation in a protein sequence. The report will evaluate the impact of the mutation on the structural domains where the residue is located, any changes on this residue, and known variants for this residue.37
Molecular modeling of native and variants of MC4R proteinWe used the 3D structure of the MC4R protein identified by El Fessikh et al. (2021).16 The 3D variant structure was generated using PyMOL version 2.4.1, and the energy minimization for the native and variant structures was achieved using the YASARA Force Field Minimization Server (http://www.yasara.org/minimizationserver.htm). The local 3D structural differences between the native and variant structures were determined using YASARA View. This graphical program for molecular modeling and simulation can produce hydrogen bonds and hydrophobic interactions between the amino acid of interest and the approximate amino acids.37
ResultsFor the identification of deleterious variants in the MC4R gene, we were able to extract a total of 1678 mutations using the NCBI dbSNP database. A total of 115 of these SNPs are found in the 3′ UTR region, 682 upstream, 348 missense, 127 synonymous, 169 in the 5′ UTR region, 193 downstream, and others.
Subsequently, only SNPs exhibiting obesity as the main clinical feature and having “pathogenic” clinical significance were selected from the ClinVar database. For the MC4R gene, 20 SNPs were selected (Appendix A, Supplementary Table 1). The selected variants include the following: D37V, V50M, S58C, E61K, N62S, I69R, D90N, N97D, I102S, S136F, R165Q, R165W, V166I, I170V, A219V, C271R, C271Y, F280L, P299H, and I316S. Fig. 1 shows a schematic representation of the MC4R protein, with the location of the mutations identified as having “pathogenic” clinical significance.
28). The selected mutations include D37V in the N-terminal, V50M, S58C, E61K, N62S and I69R in the 1st transmembrane helix (TM1), D90N, N97D, and I102S in the 2nd transmembrane helix (TM2), S136F in the 3rd transmembrane helix (TM3), R165Q, R165W, V166I, I170V in the 4th transmembrane helix (TM4), A219V in the intracellular loop 3 (ICL3), C271R and C271Y in the extracellular loop 3 (ECL3), F280L and P299H in the 7th transmembrane helix (TM7) and I316S in the C-terminal. Mutated amino acids are marked in red. ECL: extracellular loop; ICL: intracellular loop.")
Schematic representation of MC4R protein showing the 20 mutations identified as having “pathogenic” clinical significance (modified from Lotta et al. (2019)28). The selected mutations include D37V in the N-terminal, V50M, S58C, E61K, N62S and I69R in the 1st transmembrane helix (TM1), D90N, N97D, and I102S in the 2nd transmembrane helix (TM2), S136F in the 3rd transmembrane helix (TM3), R165Q, R165W, V166I, I170V in the 4th transmembrane helix (TM4), A219V in the intracellular loop 3 (ICL3), C271R and C271Y in the extracellular loop 3 (ECL3), F280L and P299H in the 7th transmembrane helix (TM7) and I316S in the C-terminal. Mutated amino acids are marked in red. ECL: extracellular loop; ICL: intracellular loop.
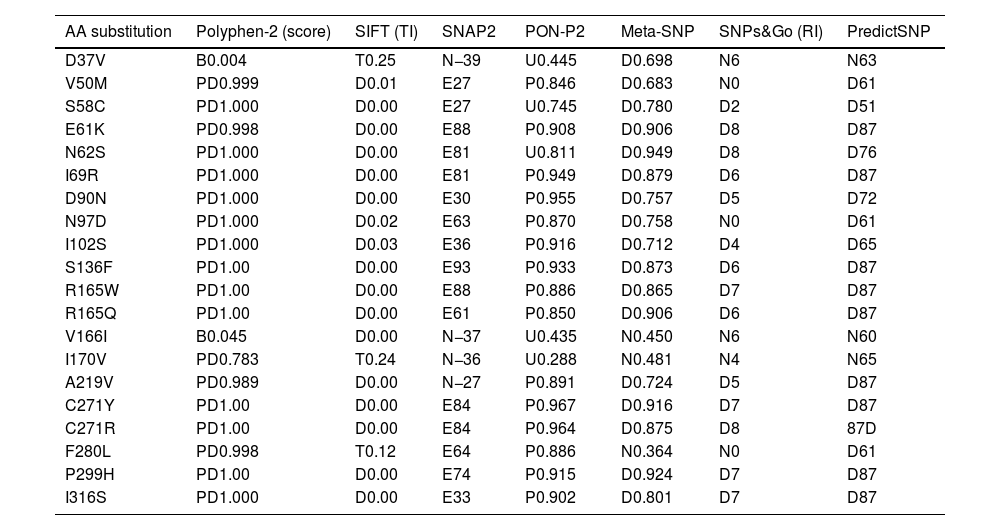
After that, 7 prediction algorithms were used to predict the functional and pathogenic impact of the 20 SNPs studied. These tools showed that most SNPs were deleterious and pathogenic, except for D37V, V166I, I170V, A219V, and F280L, which were excluded from the study (Table 1). Mutations predicted to be deleterious and pathogenic by all bioinformatics tools were selected in the study for further functional and structural analysis.
Functional and pathogenic effects of MC4R gene mutations predicted by different bioinformatics tools.
| AA substitution | Polyphen-2 (score) | SIFT (TI) | SNAP2 | PON-P2 | Meta-SNP | SNPs&Go (RI) | PredictSNP |
|---|---|---|---|---|---|---|---|
| D37V | B0.004 | T0.25 | N−39 | U0.445 | D0.698 | N6 | N63 |
| V50M | PD0.999 | D0.01 | E27 | P0.846 | D0.683 | N0 | D61 |
| S58C | PD1.000 | D0.00 | E27 | U0.745 | D0.780 | D2 | D51 |
| E61K | PD0.998 | D0.00 | E88 | P0.908 | D0.906 | D8 | D87 |
| N62S | PD1.000 | D0.00 | E81 | U0.811 | D0.949 | D8 | D76 |
| I69R | PD1.000 | D0.00 | E81 | P0.949 | D0.879 | D6 | D87 |
| D90N | PD1.000 | D0.00 | E30 | P0.955 | D0.757 | D5 | D72 |
| N97D | PD1.000 | D0.02 | E63 | P0.870 | D0.758 | N0 | D61 |
| I102S | PD1.000 | D0.03 | E36 | P0.916 | D0.712 | D4 | D65 |
| S136F | PD1.00 | D0.00 | E93 | P0.933 | D0.873 | D6 | D87 |
| R165W | PD1.00 | D0.00 | E88 | P0.886 | D0.865 | D7 | D87 |
| R165Q | PD1.00 | D0.00 | E61 | P0.850 | D0.906 | D6 | D87 |
| V166I | B0.045 | D0.00 | N−37 | U0.435 | N0.450 | N6 | N60 |
| I170V | PD0.783 | T0.24 | N−36 | U0.288 | N0.481 | N4 | N65 |
| A219V | PD0.989 | D0.00 | N−27 | P0.891 | D0.724 | D5 | D87 |
| C271Y | PD1.00 | D0.00 | E84 | P0.967 | D0.916 | D7 | D87 |
| C271R | PD1.00 | D0.00 | E84 | P0.964 | D0.875 | D8 | 87D |
| F280L | PD0.998 | T0.12 | E64 | P0.886 | N0.364 | N0 | D61 |
| P299H | PD1.00 | D0.00 | E74 | P0.915 | D0.924 | D7 | D87 |
| I316S | PD1.000 | D0.00 | E33 | P0.902 | D0.801 | D7 | D87 |
AA: amino acid; TI: tolerance index; RI: reliability index; B: benign; PD: probably deleterious; D: deleterious; E: effect; P: pathogenic; U: unknown.
Afterwards, we performed further analyses on the mutations identified as potentially damaging. Using ConSurf Server, we analyzed the degree of conservation of MC4R protein residues (Fig. 2). Based on the conservation results, 9 SNPs were located in highly conserved regions (9 on the conservation scale), 2 amino acids were predicted to be both exposed and functional (E61K, C271Y, and C271R), and 6 residues were considered buried and structural (S58C, N62S, D90N, N97D, S136F, and P299H). A total of 6 SNPs were located in conserved regions (8 on the conservation scale), 2 SNPs were predicted as exposed (R165W and R165Q), and 1 residue was predicted as buried (I69R). One SNP was located in moderately conserved regions (7 on the conservation scale) and predicted to be exposed (I316S). The 2 SNPs V50M and I102S were predicted as variable residues and were therefore not selected for further analysis.
.")
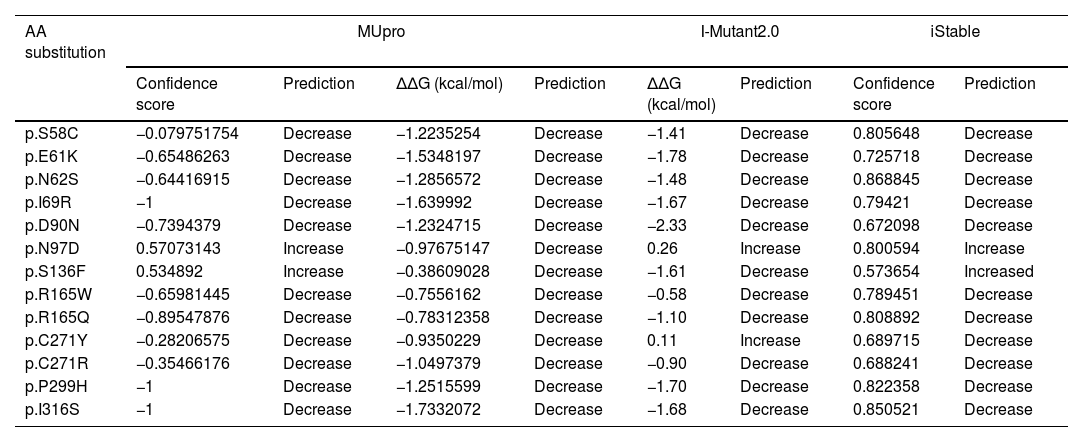
To study the effect of variants on MC4R protein stability, MUpro, I-Mutant2.0, and iStable tools were used (Table 2). According to the results, most studied SNPs decreased protein stability except for N97D, S136F, and C271Y. According to MUpro, I-Mutant2.0, and iStable servers, the variants S58C, E61K, N62S, I69R, D90N, R165Q, P299H, and I316S are the SNPs that reduce protein stability the most. These variants are selected for further analysis.
Prediction of change in protein stability using MUpro, I-Mutant2.0, and iStable.
| AA substitution | MUpro | I-Mutant2.0 | iStable | |||||
|---|---|---|---|---|---|---|---|---|
| Confidence score | Prediction | ΔΔG (kcal/mol) | Prediction | ΔΔG (kcal/mol) | Prediction | Confidence score | Prediction | |
| p.S58C | −0.079751754 | Decrease | −1.2235254 | Decrease | −1.41 | Decrease | 0.805648 | Decrease |
| p.E61K | −0.65486263 | Decrease | −1.5348197 | Decrease | −1.78 | Decrease | 0.725718 | Decrease |
| p.N62S | −0.64416915 | Decrease | −1.2856572 | Decrease | −1.48 | Decrease | 0.868845 | Decrease |
| p.I69R | −1 | Decrease | −1.639992 | Decrease | −1.67 | Decrease | 0.79421 | Decrease |
| p.D90N | −0.7394379 | Decrease | −1.2324715 | Decrease | −2.33 | Decrease | 0.672098 | Decrease |
| p.N97D | 0.57073143 | Increase | −0.97675147 | Decrease | 0.26 | Increase | 0.800594 | Increase |
| p.S136F | 0.534892 | Increase | −0.38609028 | Decrease | −1.61 | Decrease | 0.573654 | Increased |
| p.R165W | −0.65981445 | Decrease | −0.7556162 | Decrease | −0.58 | Decrease | 0.789451 | Decrease |
| p.R165Q | −0.89547876 | Decrease | −0.78312358 | Decrease | −1.10 | Decrease | 0.808892 | Decrease |
| p.C271Y | −0.28206575 | Decrease | −0.9350229 | Decrease | 0.11 | Increase | 0.689715 | Decrease |
| p.C271R | −0.35466176 | Decrease | −1.0497379 | Decrease | −0.90 | Decrease | 0.688241 | Decrease |
| p.P299H | −1 | Decrease | −1.2515599 | Decrease | −1.70 | Decrease | 0.822358 | Decrease |
| p.I316S | −1 | Decrease | −1.7332072 | Decrease | −1.68 | Decrease | 0.850521 | Decrease |
AA: amino acid; ΔΔG: delta delta G.
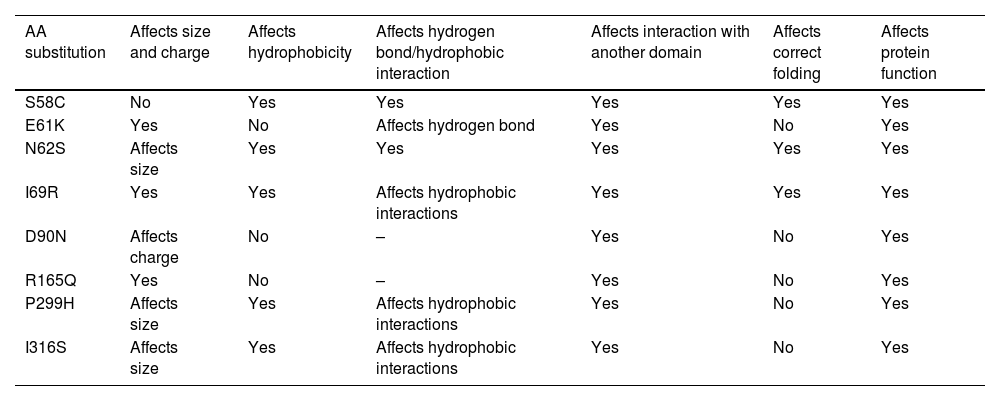
According to HOPE analysis, each amino acid has its specific size, charge, and hydrophobicity value. For the N62S, I69R, D90N, and R165Q mutations, the wild-type residues lose their original charges: R165Q changes from positive to neutral, D90N from negative to neutral, N62S from negative to positive, and I69R from neutral to positive. These changes are predicted to affect the interactions of the protein with other molecules. The E61K, N62S, I69R, R165Q, P299H, and I316S mutations lead to size differences between the wild-type and mutant residues, potentially disrupting interactions with other molecules or parts of the protein. For S58C, N62S, and R165Q, the mutant residues are more hydrophobic than the wild-type residues, whereas, for I69R, P299H, and I316S, the wild-type residues are more hydrophobic than the mutants. These differences in hydrophobicity could affect the formation of hydrogen bonds and hydrophobic interactions. Additionally, the S58C and N62S mutations are expected to cause a loss of hydrogen bonds at the core of the protein, thus disrupting proper folding.
All mutated residues are situated within a pivotal domain crucial for the protein function and interact with another essential domain. The critical domain connectivity affected by the mutation could significantly impact the protein's overall function. Table 3 illustrates the HOPE analyses.
The effect of the studied mutations on the structural features of the protein determined by the HOPE server.
| AA substitution | Affects size and charge | Affects hydrophobicity | Affects hydrogen bond/hydrophobic interaction | Affects interaction with another domain | Affects correct folding | Affects protein function |
|---|---|---|---|---|---|---|
| S58C | No | Yes | Yes | Yes | Yes | Yes |
| E61K | Yes | No | Affects hydrogen bond | Yes | No | Yes |
| N62S | Affects size | Yes | Yes | Yes | Yes | Yes |
| I69R | Yes | Yes | Affects hydrophobic interactions | Yes | Yes | Yes |
| D90N | Affects charge | No | – | Yes | No | Yes |
| R165Q | Yes | No | – | Yes | No | Yes |
| P299H | Affects size | Yes | Affects hydrophobic interactions | Yes | No | Yes |
| I316S | Affects size | Yes | Affects hydrophobic interactions | Yes | No | Yes |
AA: amino acid.
Using YASARA View software, the 3D structure of the MC4R protein and its mutations (S58C, E61K, N62S, I69R, D90N, R165Q, P299H, and I316S) were compared (Fig. 3).
. E61K causes a gain of 1 hydrophobic interaction with Cys 57. N62S causes a loss of 1 hydrogen bond with Ser 295 and 1 hydrophobic interaction with Met 91. I69R causes a loss of 1 hydrogen bond and 1 hydrophobic interaction with Val 65. D90N causes a loss of 1 hydrophobic interaction with Ser 295. R165Q causes a gain of 1 hydrogen bond with Met 161. P299H causes a loss of 2 hydrophobic interactions (1 with Glu 61 and 1 with Val 65) and a gain of 1 hydrogen bond with Ser 295). I316S causes a loss of 1 hydrogen bond with Cys 319.")
The 3D structure effect of MC4R mutations. S58C causes a loss of the hydrogen bond with Ser 295 and the 2 hydrophobic interactions (one with Ser 94 and 1 with Ser 295). E61K causes a gain of 1 hydrophobic interaction with Cys 57. N62S causes a loss of 1 hydrogen bond with Ser 295 and 1 hydrophobic interaction with Met 91. I69R causes a loss of 1 hydrogen bond and 1 hydrophobic interaction with Val 65. D90N causes a loss of 1 hydrophobic interaction with Ser 295. R165Q causes a gain of 1 hydrogen bond with Met 161. P299H causes a loss of 2 hydrophobic interactions (1 with Glu 61 and 1 with Val 65) and a gain of 1 hydrogen bond with Ser 295). I316S causes a loss of 1 hydrogen bond with Cys 319.
As results for the S58C, we found that Ser58 forms 4 hydrogen bonds (1 with Leu54, 2 with Asn62, and 1 with Ser295) and 2 hydrophobic interactions (1 with Ser94 and 1 with Ser295). The substitution of Ser by Cys at codon 58 (S58C) involves the loss of the hydrogen bond with Ser295 and the 2 hydrophobic interactions. For the E61K, Glu61 forms 2 hydrogen bonds (1 with Ile57 and 1 with Val65) and 1 hydrophobic interaction with Pro299. When replaced by Lys at position 61 (E61K), 1 hydrophobic interaction with Ile57 is gained. In N62S, Asn62 forms 4 hydrogen bonds (2 with Leu59, 1 with Ile66, and 1 with Ser295). The substitution of Asn by Ser at codon 62 (N62S) involves the loss of 1 hydrogen bond with Ser295 and 1 hydrophobic interaction with Met91. For I69R, Ile69 forms 1 hydrogen bond with Val65 and 2 hydrophobic interactions (1 with Val65 and 1 with Tyr80). In the variant case, the substitution of Ile by Arg at codon 69 (I69R) involves the loss of the hydrogen bond and 1 hydrophobic interaction with Val65.
In the case of D90N, Asp90 forms 3 hydrogen bonds (1 with Leu86 and 2 with Ser94) and 1 hydrophobic interaction with Ser295. When replaced by Asn at codon 90 (D90N), the hydrophobic interaction is lost. For R165Q, Arg 165 forms 1 hydrogen bond with Ile 169. The substitution of Arg by Gln at codon 165 (R165Q) involves the gain of 1 hydrogen bond with Met161. Regarding P299H, Pro299 forms 1 hydrogen bond with Ala303 and 2 hydrophobic interactions (1 with Glu61 and 1 with Val65). The substitution of Pro by His at position 299 (P299H) involves the loss of the 2 hydrophobic interactions and a gain of 1 hydrogen bond with Ser295. In I316S, Ile316 forms 2 hydrogen bonds (1 with Thr312 and 1 with Cys319) and 1 hydrophobic interaction with Leu64. Conversely, the substitution of Ile by Ser at codon 316 (L316S) results in the loss of 1 hydrogen bond with Cys319.
The results further confirmed the deleterious effects on the function and structure of the MC4R receptor for the variants S58C, E61K, N62S, I69R, D90N, R165Q, P299H, and I316S.
DiscussionThis study aimed to evaluate the different mutations of the MC4R gene previously identified in individuals with obesity and identify the most deleterious variants from functional and structural perspectives. In fact, the MC4R gene plays a crucial role in regulating food intake. It is therefore an important model for studying genetic mutations affecting various protein domains linked to the obesity phenotype.
A total of 348 out of 1678 SNPs analyzed were missense mutations, with 20 SNPs being significantly linked to obesity as a primary clinical feature and categorized as having “pathogenic” clinical significance in the ClinVar database. Afterwards, mutations predicted to be deleterious and pathogenic by all bioinformatics tools were selected in the study for further functional and structural analysis. These variants include the following: S58C, E61K, N62S, and I69R in the TM1, D90N in the TM2, R165Q in the TM4, P299H in the TM7, and I316S in the C-terminal. These mutations have been identified in subjects with severe early-onset obesity.38–45
Functional studies indicate that the S58C, E61K, N62S, I69R, R165Q, P299H, and I316S variants strongly diminish cell surface expression, reducing it by approximately ≥60% vs the wild-type receptor.38,46 These mutations cause misfolding of the MC4R receptor, leading to its retention in the endoplasmic reticulum and subsequent ubiquitination. This results in a significantly reduced receptor cell surface expression and activation by α-MSH, leading to impaired cAMP ERK1/2 signaling and contributing to obesity.38,47,48 In the other case, the D90N mutation shows normal cell surface expression; however, it leads to a complete loss of cAMP production.49 This mutation is situated distantly from the binding pocket and is likely involved in conformational changes crucial for MC4R activation.50
In our analysis, the mutated residues are situated in a domain pivotal for the protein activity and lead to disruptions in hydrogen bonding and/or hydrophobic interactions with other domains, potentially affecting the receptor's conformation and activation. The loss of contact between TM1 and TM7 (S58C, N62S, and P299H) might destabilize the active state of MC4R, leading to a reduced affinity for α-MSH. The critical role of TM7 in cell surface expression and effector activation of MC4R has been shown in previous studies. It has been found that disruption of contact between TM1 and TM7 could lead to structural instability and misfolding during receptor activation, suggesting the importance of these interactions in functional receptor dynamics.38,50–52 Furthermore, reduced interactions between TM1 and TM2 (S58C and N62S) may lead to the structural flexibility of ICL1 and, consequently, affect MC4R folding and trafficking.38 In addition, the loss of interaction between TM2 and TM7 (D90N) results in a receptor state that cannot be fully activated by ligand action.53 In fact, the D90N mutation does not affect the receptor's cell surface expression but does result in a complete loss of cAMP production following stimulation by α-MSH, which indicates a loss of signal transduction mediated by Gs/AC activation.49,54 Normally, ligand binding to MC4R triggers a conformational change that allows interaction with G proteins such as Gs, initiating the cAMP-PKA-CREB signaling pathway leading to increased c-Fos transcription and decreased AMPK phosphorylation and activity.7,8 In the case of I316S, the loss of an interaction between the C-terminal and H8 segment could affect the protein function. Indeed, H8 plays multiple roles in receptor internalization, dimerization, and interaction with G proteins and arrestin.55,56 Additionally, the amino acids from Lys314 to Cys319 are essential for proper receptor function, particularly for their correct localization on the cell surface. Indeed, this region truncation results in impaired cell surface localization and disrupted signaling.57
On the other side, some mutations (E61K, R165Q, and P299H) induce the gain of new interactions, potentially influencing receptor conformation. Notably, the formation of hydrogen bonds/hydrophobic interactions might stabilize an incorrect protein conformation and lead to a defect in receptor activation.38,58 It is widely recognized that hydrophobic interactions contribute significantly to protein stability (E61K).58 Furthermore, the formation of a new hydrogen bond between TM4 and ICL2 (R165Q) may stabilize the receptor and compromise coupling with G proteins and β-arrestin involved in the trafficking of MC4R leading to its intracellular retention. In fact, research suggests that the ICL2 may be essential for receptor activation and desensitization.38 Thus, these alterations in hydrogen bonds and/or hydrophobic interactions might destabilize the MC4R receptor affecting signaling pathways.
These findings highlight the critical role of specific MC4R mutations in the pathogenesis of obesity and provide insights into the structural and functional consequences of these genetic variations.
Limitations- -
The study analyzed missense variations in the MC4R gene in silico, which may not fully represent actual biological effects in living systems.
- -
Computational tools were used to predict deleterious and pathogenic SNPs; however, these tools have limitations in accurately assessing the impact on protein function.
- -
In silico studies are an important step, but experimental studies (in vitro and in vivo studies), must complement them to validate the results and fully understand their impact on obesity.
Numerous mutations within the MC4R gene lead to either partial or complete loss of function. These mutations have been linked to structural alterations in the receptor, preventing it from undergoing the necessary conformational changes crucial for protein activation and subsequent signal transduction.
Authors’ contributionsMeriem El Fessikh: Study concept and design, data acquisition, data analysis, data interpretation, and manuscript writing. Hanaa Skhoun: Study concept and design, and manuscript revision. Zohra Ouzzif: Manuscript revision. Jamila El Baghdadi: Study concept and design, data interpretation, and manuscript revision. All authors have read and agreed to the published version of the manuscript.
Ethics approvalNot applicable.
Consent to participateNot applicable.
Consent for publicationNot applicable.
FundingNone declared.
Conflicts of interestNone declared.
Data availability statementData sharing is not applicable to this article as no new data were generated.
The followings are the supplementary data to this article:
