






There are some well known factors involved in the etiology of thyroid cancer, including iodine deficiency, radiation exposure at early ages, or some genetic changes. However, epigenetic modulators that may contribute to development of these tumors and be helpful to for both their diagnosis and treatment have recently been discovered. The currently known changes in DNA methylation, histone modifications, and non-coding RNAs in each type of thyroid carcinoma are reviewed here.
Son conocidos algunos factores implicados en la etiología del cáncer de tiroides como el déficit de yodo o la exposición a radiación en edades tempranas o algunas alteraciones genéticas. Sin embargo, en los últimos años se han descubierto moduladores epigenéticos que puedan contribuir al desarrollo de estos tumores y podrían tener una utilidad tanto en el diagnóstico como en el tratamiento. En esta revisión se repasan las alteraciones conocidas hasta ahora tanto en la metilación del ADN como en las modificaciones de las histonas y los ARN no codificantes en cada uno de los tipos de carcinomas de tiroides.
Thyroid carcinoma is usually divided into three histological classes: differentiated tumors and non-differentiated tumors, the latter being further subdivided into anaplastic thyroid cancer (ATC) and medullary thyroid carcinoma (MTC). The differentiated group represents 90% of diagnosed tumors, the most frequent being papillary thyroid cancer (PTC) (80–85%), followed by follicular thyroid carcinoma (FTC, 10–15%).1–3
The etiology of thyroid cancer involves factors such as exposure to external radiation, living in iodine deficit regions, a family history of thyroid cancer, or being female, although the underlying mechanisms involved in this process are still not fully understood.4 The genetic alterations described in thyroid cancer development include BRAF (B-raf-protooncogen-serine/threonine kinase) gene mutations, especially BRAFV600E, which leads to the constitutive activation of kinase and the chronic stimulation of MAP-kinase (mitogen-activated protein kinase) signaling. A single nucleotide mutation is present in nearly 50% of PTC tumors and 23% of ATCs, but is not present in FTCs and benign thyroid nodules.5 The BRAF mutation seems to be a specific marker for the aggressive phenotype of PTC and ATC tumors,6 due to its role in angiogenesis, extracellular matrix alteration and promotion of tumor invasion.7 However, the work developed by George et al.8 described the role of the TERT (telomerase reverse transcriptase) promoter mutation in the survival rate of PTC patients, while the BRAF mutation was found to have no effect. This seems to contradict previously published data, but it may be that the small number of patients analyzed in this work who did not have the BRAF-mutation makes the comparison of results less reliable.9
RAS family mutations are frequently associated with FTC. They lead to the constitutively activated GTP-bound state of this protein, which stimulates the PI3K–AKT (phosphatidylinositol-3-kinases-AKT serine/threonine kinase 1) pathway. A change which is associated with less favorable FTC prognosis.10 RET (rearranged during transfection) translocation is a genetic alteration linked to thyroid cancer development. RET encodes a tyrosine kinase receptor that activates the MAPK and PIK3-AKT cascades. RET rearrangements are common in 98% of hereditary and 40% of sporadic forms,11 but are also frequent in radiation-associated thyroid cancer and in PTC developed at early ages.12
Another common gene rearrangement is the PAX8–PPARG fusion gene (paired box 8-peroxisome proliferator activated receptor-γ). It is originated by a translocation (t(2;3) (q13;p25)) that interferes with PPARG activity or acts as a PPARG-like transcription factor and is present in nearly 40% of FTCs, meaning that it can be used as a diagnostic marker to determine the most suitable treatment of positive tumors. In contrast, it is present in less than 1% of PTCs.13
Complex diseases such cancer cannot, however, be explained as resulting from a simple genetic mutation or by special environmental influences, rather in certain environmental circumstances the epigenetic and the genetic, acting as independent mechanisms, contribute to cancer development.
Epigenetics was first described by Conrad Waddington in 194214 as the scientific study of the interaction between genes and the environment which leads to a phenotype. This discipline analyzes the inherited changes experienced by genetic material which are not due to alterations in the sequence of the DNA.15 A more detailed description of the epigenetic mechanisms involved in cancer will be provided in the following sections.
This review will discuss the role of DNA methylation, histone modifications and non-coding RNAs on thyroid cancer development, placing emphasis on the advantages of identifying these mechanisms in order to design the best therapeutic strategies.
Epigenetic modifications related to cancerDNA methylationGenerally, tumoral cells are characterized by an aberrant DNA methylation pattern defined by a global loss of methylation (global hypomethylation) which is frequently located in repetitive transposable elements, gene bodies and intergenic regions, as well as an increase in methylation in specific regions (gene promoters). Moreover, DNA methylation can create an enabling environment for gene mutations to develop, which would also contribute to tumor progression.16
In mammals, DNA methylation consists in the addition of a methyl group in the 5′ position of cytosines that precede guanines, called CpG dinucleotides. Those sites are frequently concentrated in regions known as CpG islands (Fig. 1). During DNA replication, these methylated cytosines can undergo spontaneous deamination and be transformed into thymines. If this deamination process is experienced by a demethylated cytosine, it will be converted into uracil. These changes (cytosine deamination) in the double DNA strand, originate G:T or G:U, both of which will result in the conversion of a C to a T in the new strand. Specific enzymes such as TDG (thymine DNA glycosylase) and MBD4 (methyl-CpG-binding protein 4) are able to repair these incorrect matches,17 while these positions are hot spots for the generation of spontaneous transitions observed in cancer and other genetic diseases.18
 of the target mRNA, causing the inhibition of the translational process, or mRNA fragmentation and ultimately, gene silencing or chromatin remodeling.")
Epigenetic mechanisms. DNA methylation, covalent addition of a methyl group to cytosine within CpG dinucleotides is mediated by DNA methyltransferases. Unmethylated CpGs within promoter regions do not abolish transcription and there is gene expression. When methylation occurs, the genes become silenced. Histone modifications, one of the histone changes, acetylation at the lysine residue, neutralizes its positive charge, resulting in a weakened interaction between histone and DNA. The chromatin acquires a relaxed conformation that allows gene transcription. mi-RNA, micro-RNAs form hairpin structures with the 3′ untranslated region (3′-UTR) of the target mRNA, causing the inhibition of the translational process, or mRNA fragmentation and ultimately, gene silencing or chromatin remodeling.
Global hypomethylation was first described in colon cancer by Feinberg et al.19 Loss of methylation in CpG islands associated with gene promoters can lead to the restoration of oncogenes or the genes involved in various features of cancer.20 In addition, this hypomethylation also affects satellite sequences, repetitive genome sequences (i.e. LINEs (long interspersed nuclear elements) and SINEs (short interspersed nuclear elements)) and the transposable elements that lead to chromosomal instability linked to tumor development.21
DNA hypomethylation of specific genes in tumoral cells is a rare event, but some examples related with tumor stage or treatment response have been found. Hypomethylation of JAG1 and NOTCH 1 is related to lymph node metastasis and the advanced stages of breast cancer.22 In addition, platinum chemotherapy resistance of high grade serous ovarian cancer has been associated with the hypomethylation of the homeobox gene MSX1 (Msh homeobox 1) involved in cellular differentiation.23
Hypermethylation of CpG islands in tumor suppressor genes, frequently associated with gene silencing, has been widely studied in cancer. This includes the genes involved in biological processes such as cell cycle, DNA repair, immune response, cell signaling, apoptosis, angiogenesis and cancer metastasis.24
Histone modificationsHistone proteins are part of the nucleosome. This structure is the fundamental repeating unit of chromatin, consisting of a protein octamer containing two molecules of each core histone (H2A, H2B, H3 and H4) which are small and extremely alkaline, wrapped around by the 147 base pairs of genomic DNA.
Histones also have some amino-terminal tails which extend freely from the DNA-protein octamer, making them open to the modification of their amino acid residues. Post-translational modifications on histone tails include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, biotination, citrullination, poly-ADP-ribosylation, and N-glycosylation. The diverse number of combinations that can occur between several of these changes has culminated in the elaboration of the “histone code”.25
Histone modifications are the result of the balance between different groups of enzymes, some with antagonist activity (Fig. 1). For example, histone acetylation is catalyzed by histone acetyltransferases (HATs), and the reverse action by histone deacetylases (HDACs), while the enzymes involved in histone methylation are the substrate specific HMTs (histone methyl transferases) and the antagonistic HDMs (histone demethylases).26 Other enzymes involved in histone modification are ubiquitin ligases, histone phosphatases, and glycohyrolases. Their specific roles in tumor development are, however, not fully documented and further research is necessary to elucidate this.27
The main functions of the histones are in establishing the structural domains of chromatin and managing transcription, as well as the replication and repair of DNA and chromosome condensation. Modification of histones can induce changes between them and the DNA, or they can act as binding sites for the recruitment of other proteins that recognize these changes.28 In general, acetylation of lysines favors transcriptional activity; the addition of acetyl groups neutralizes the positive charge of the lysines and reduces their affinity for the DNA, which facilitates access to the transcriptional machinery. Methylation can occur in lysine and arginine residues, which involve between one and three methyl groups, the effect of this modification depending on the residue concerned and the degree of methylation.29
Aberrant HAT and HDAC activity are both associated with cancer development. Yang demonstrated the role of HATs such as p300 in hematological tumor development,30 and HDAC overexpression has been described in solid tumors such as prostate, renal and breast tumors.31
Non-coding RNAsNon-coding RNAs have been recently linked to the development, progression and diagnosis of cancer. They have been recognized as important regulators of gene expression involved in heterochromatin development and gene silencing at the transcriptional and post-transcriptional levels. They can be classified as follows: small interfering RNAs (siRNAs), micro RNAs (miRNAs), piwi associated RNAs (piRNAs), long ncRNAs (lncRNAs) and enhancer RNAs (eRNAs).32–35
miRNAs are the best characterized form of non-coding RNA. They are endogenous molecules of 19–24 nucleotides of non-coding RNA which form hairpin structures with the 3′ untranslated region (3′-UTR) of the target mRNA, causing the inhibition of the translational process or mRNA fragmentation, and, ultimately, gene silencing (Fig. 1). The miRNAs control a number of different processes in the cell, such as differentiation, proliferation, and at least 60% of the genes encoding proteins are subject to regulation by miRNAs. During cancer development, miRNA expression is anomalously regulated, which can alter the expression of cancer-related genes and result in the lack of regulation of cellular pathways.36
lnc RNAs are longer than 200 nucleotides, and recently have been related to cancer development. The action mechanism, while not fully understood, is known to involve them interacting with and inducing changes in chromatin, or acting as antisense transcripts.37
Epigenetic marks involved in thyroid cancerDNA methylation in thyroid cancerOur group previously described for the first time the genome wide promoter methylation status of papillary, follicular, medullary and anaplastic thyroids tumors as well as non-tumorigenic thyroid tissues.38 With respect to the epigenetic marker of methylation, differential methylation patterns were identified for each tumor subtype analyzed.
In general, higher hypermethylation was found in differentiated thyroid tumors compared to healthy samples, while non-differentiated tumors were preferentially hypomethylated. These results were later widely extended with the works developed by Mancikova et al.39 and Ellis et al.40 on the well-differentiated variants. The data obtained in these studies allowed the identification of differential methylation patterns not only between the benign forms, such as between FAs and the PTC and FTC forms, but also between PTCs and FVPTCs (follicular variant of papillary thyroid carcinomas). FTC carcinomas were found to have a higher methylation profile, and this aberrant methylation profile seems to be related to tumor progression. Moreover, an association was found between the presence of the BRAF and RAS mutations and RET/PTC rearrangements and the appearance of altered methylation patterns when compared with tumoral forms without these mutations. These differentially methylated CpGs were linked with genes such as NIS (sodium-iodide symporter), RARβ2 (retinoid acid receptor β2) and TIMP3 (tissue inhibitor metallopeptidase 3), all of which are involved in cellular proliferation and metastasis, suggesting a role for the BRAF mutation in tumor progression and the more aggressive behavior of PTC tumors.40
The potential role in cancer development was described for the hypermethylated HOXB4 and ADAMTS8 in the PTC variant and the hypermethylation of ZIC1 and KISS1R in FTCs, was extended by Mancikova et al. to incorporate the COL4A2 and DLEC1 genes in these variants as well as them observing hypomethylation in the KLK10 gene, which is strongly associated with the BRAF mutation positive PTC variants.39 All these genes have been identified as having tumor suppression activity in cancer, except the KLK10 gene, which encodes a protein involved in extracellular matrix degradation. The differential methylation pattern observed in these genes compared with normal tissue or the benign variants suggests their role in tumor progression. Moreover, KISS1R (GPR54) function in thyroid tumors was described by Savvidis et al.,41 who observed its reduced expression in invasive differentiated thyroid tumors, which concurs with the promoter methylation we observed in FTC.
In contrast, we observed aberrant hypomethylation in undifferentiated variants. We found promoter hypomethylation of the NOTCH4 gene in ATC. This gene is overexpressed in thyroid tumors compared with healthy samples and could be involved in tumor angiogenesis.42 Recently, its role in primary glioblastoma angiogenesis has been described,43 as well as in gastric cancer growth promotion,44 suggesting a similar role to the one it has in the aggressive forms of thyroid cancer.
Other genes under the control of methylation include the phosphatase and tensin homolog gene (PTEN) which has a tumor suppression function, through its antagonism of the PI3K/Akt pathway, and which has been described hypermethylated in differentiated thyroid tumors.45 Recent works developed by Ng et al.46 demonstrated increased methylation of PTEN in blood samples of thyroid and breast cancer patients. In addition, loss of PTEN expression was also observed in FVPTC tumors, but it was not associated with gene deletion, and for this reason the authors proposed that methylation was the cause of this lack of expression.47 The RAS association domain family protein 1 (RASSF1A) regulates RAS protein function and is involved in cell cycle regulation and the mitotic process. RASSF1A promoter hypermethylation has been found to be an early event in tumor development in PTC and in follicular thyroid hyperplasia.48,49
An additional recently identified tumor suppressor gene is RASAL1 (Ras protein activator like 1). This gene has GTPase activity and is involved in RAS signaling. RASAL1 has been found hypermethylated in nearly 27% of FTCs and 17% of ATCs, supporting the notion that it has a role in the development of these types of thyroid tumors.50 In addition, Wang et al.51 have demonstrated that TERT (telomerase reverse transcriptase) up-regulation results from gene promoter methylation. The role of TERT as an oncogene has been previously described.52 Wang et al. suggested that methylation causes the dissociation of repressor proteins from their binding sequences, and that this causes gene activation and telomere preservation in this class of thyroid tumors.
REC8 is a tumor suppressor gene found hypermethylated in thyroid cancer and has been correlated with poor prognosis. This gene exerts its effect through the PIK3 pathway, while its inactivation can lead to oncogenic development by altering this route.53GPX3, another tumor suppressor gene, is also a candidate in thyroid cancer development. It has been described hypermethylated in the promoter region in nearly 50% of PTC samples. This epigenetic change alters the Wnt/beta-catenin pathway facilitating the progression of metastasis.54 Papillary thyroid tumor relapse is associated with the methylation status of the RUNX3 gene. This protein belongs to a family of transcription factors, RUNX, and has been identified as having a tumor suppressor function through its modulation of apoptosis and cell proliferation in solid tumors. The association between RUNX3 methylation and PTC recidivism has led to this gene becoming a potential candidate for the treatment of PTC patients.55 It is important to highlight the work developed by Agrawal et al.56 These authors developed the genomic landscape of 496 PTC samples, enabling the identification of different profiles (at the genetic, epigenetic and proteomic level) of PTC tumors in relation to BRAF and RAS mutations. These differences could facilitate improvements in and the personalization of therapies against these tumors.
Histone modifications in thyroid cancerAggressive forms of thyroid cancer are frequently resistant to radioactive iodine therapy and the use of histone deacetylase (HDAC) inhibitors shows great promise for the treatment this type of tumor.57 The anticancer effect of HDAC inhibitors, whether in combination with other antitumor agents or not, leads to the growth of cancer cells being minimized, as well as increasing the radioiodine uptake of tumoral cells.
Jang et al.58 have revealed the benefits of treating metastatic FTC and ATC cell lines with HDAC inhibitors. These enzymes control the acetylation/deacetylation levels of chromatin. In the study cited, cell lines were treated with a set of thirteen HDAC inhibitors that arrested cell growth and induced apoptosis, increased levels of caspase-3 and PARP proteins, as well as of CDK/cyclin proteins which act as cell cycle checkpoints. These results confirmed those previously obtained by Mitmaker et al. who combined this therapy with a demethylating agent, 5-azacytidine (5-AZC).59 Treatment of anaplastic thyroid tumor cell lines with thailandepsin A (TDP-A), another HDAC, caused an increase in caspase and CDK/cyclin inhibitors in these cell lines.60 Similar results have been obtained with another HDAC inhibitor, N-hydroxy-7-(2-naphthylthio) hepatonomide (HNHA), in ATC and PTC cell lines and in mice models. Treatment with this drug raised p21 levels, a pro-apoptotic protein, and reversed gene silencing by increasing histone acetylation.61
The role of PXD-101 on ATC cell lines has been previously described by Lin et al.,62 who demonstrated that PXD-101 caused cell cycle arrest and apoptosis in transformed cells due to a reduction in thioredoxin activity and the inhibition of RAS/RAF/ERK and PI3K/AKT/mTOR pathways. These results were confirmed by Kim et al.63 with the combination of PXD-101 and a heat shock 90 protein inhibitor (AUY922).
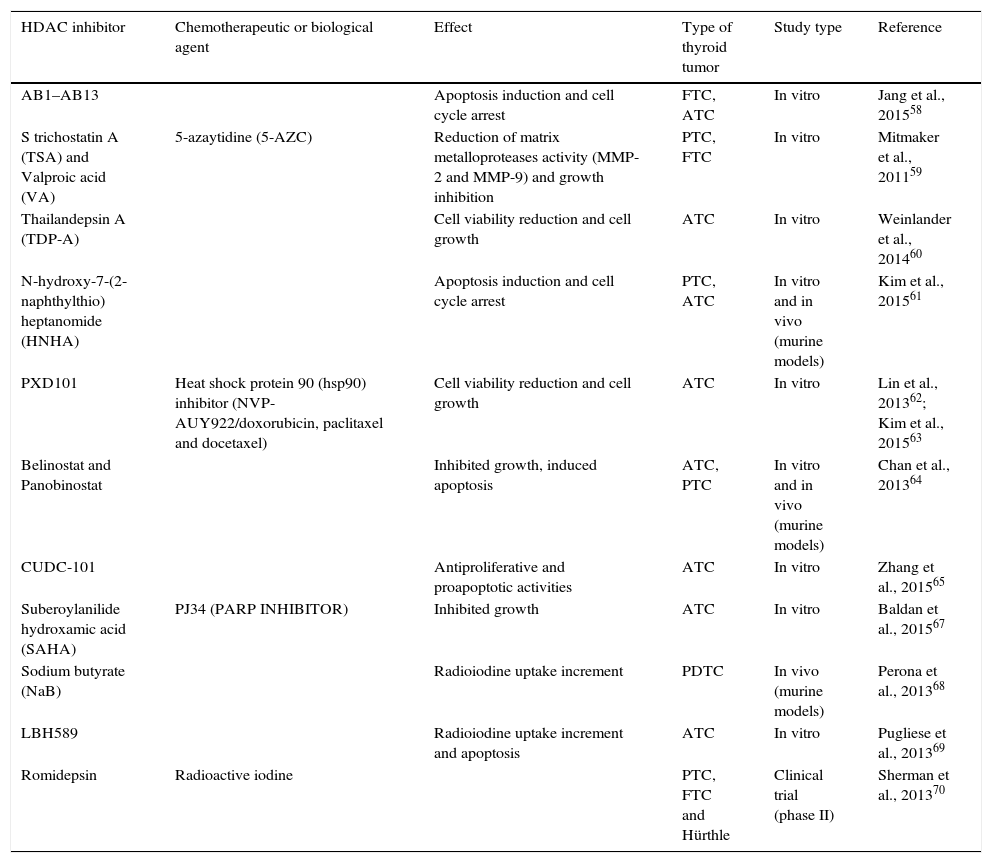
BDR4 is a bromodomain protein frequently upregulated in thyroid cancer tissues. Inhibition of BDR4, suppressed tumor growth, indicating that this protein likely has a role in tumor progression. BDR4 protein binds to acetylated histones and facilitates the recruitment of transcription factors, and its chaperone function has meant that this protein is considered a promising chemotherapeutic agent.64 A summary of clinical trials and in vitro studies is detailed in Table 1.65–70
Summary of histone deacetylase inhibitor trials in thyroid cancer.
| HDAC inhibitor | Chemotherapeutic or biological agent | Effect | Type of thyroid tumor | Study type | Reference |
|---|---|---|---|---|---|
| AB1–AB13 | Apoptosis induction and cell cycle arrest | FTC, ATC | In vitro | Jang et al., 201558 | |
| S trichostatin A (TSA) and Valproic acid (VA) | 5-azaytidine (5-AZC) | Reduction of matrix metalloproteases activity (MMP-2 and MMP-9) and growth inhibition | PTC, FTC | In vitro | Mitmaker et al., 201159 |
| Thailandepsin A (TDP-A) | Cell viability reduction and cell growth | ATC | In vitro | Weinlander et al., 201460 | |
| N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA) | Apoptosis induction and cell cycle arrest | PTC, ATC | In vitro and in vivo (murine models) | Kim et al., 201561 | |
| PXD101 | Heat shock protein 90 (hsp90) inhibitor (NVP-AUY922/doxorubicin, paclitaxel and docetaxel) | Cell viability reduction and cell growth | ATC | In vitro | Lin et al., 201362; Kim et al., 201563 |
| Belinostat and Panobinostat | Inhibited growth, induced apoptosis | ATC, PTC | In vitro and in vivo (murine models) | Chan et al., 201364 | |
| CUDC-101 | Antiproliferative and proapoptotic activities | ATC | In vitro | Zhang et al., 201565 | |
| Suberoylanilide hydroxamic acid (SAHA) | PJ34 (PARP INHIBITOR) | Inhibited growth | ATC | In vitro | Baldan et al., 201567 |
| Sodium butyrate (NaB) | Radioiodine uptake increment | PDTC | In vivo (murine models) | Perona et al., 201368 | |
| LBH589 | Radioiodine uptake increment and apoptosis | ATC | In vitro | Pugliese et al., 201369 | |
| Romidepsin | Radioactive iodine | PTC, FTC and Hürthle | Clinical trial (phase II) | Sherman et al., 201370 |
ATC: anaplastic thyroid cancer; FTC: follicular thyroid cancer; PTC: papillary thyroid cancer; PDTC: poorly differentiated thyroid carcinoma; HDAC: histone deacetylase.
Each thyroid tumor subtype seems to have a specific mRNA methylation pattern that leads to specific diagnosis, treatment response or prognosis of the tumor (Table 2).71 Among the different subtypes of non-coding RNAs, the miRNAs are probably the most widely described group. They can act as oncogenes or tumor suppressor genes, controlling apoptosis, cell cycle and angiogenesis, all functions involved in cancer progression.72 However, only a small number of miRNAs have been properly identified as having a potential role in diagnosis or prognosis in thyroid cancer. A brief summary of the non-coding RNAs involved in thyroid cancer is included in Table 2.
Summary of epigenetic mechanisms in thyroid cancer.
| Type of thyroid tumor | Gene methylation | Effect | Histone modifications | Effect | mi RNA (<200 nt) | Effect | Long non-coding RNAs (lncRNAs) (>200 nt) | References | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypermethylation | Hypomethylation | Up-regulated | Down-regulated | Up-regulated | Down-regulated | ||||||
| PTC | HOXB4, ADAMTS8, RARB2, TIMP3, DLEC1, COL4A2, NIS, PTEN, RASSF1, RUNX3, REC8, GPX3 | KLK10 | Metastasis, cellular proliferation, activation matrix metalloproteases activity | Acetylation | Apoptosis reduction and inhibition cell cycle arrest | miR-146b-5p, miR-1244, miR-127-3p, miR-128, miR-130b, miR-134, miR-135b, miR-139, miR-141, miR-144,miR-146, miR-155, miR-15a, miR-16, miR-164b, miR-17, miR-172, miR-181, miR187,miR191, miR-1975, miR-199, miR-200, miR-203, miR-205, miR-21, miR-210, miR-213, miR-214, miR-220, miR-223, miR-224, miR-31, miR-32, miR-342-3p, miR-34a, miR-375,miR-543, miR-551b,miR-720,miR-768-3p,miR-7e, miR-81a, miR-99b-3p, mi-1274a, mi-551b | let-7, miR-1, miR-7-5p, miR-1179, miR-1225-5p, miR-122-5p, miR-1231, miR-126, miR-1268, miR-1278, miR-130,miR-137, miR-138, miR-140, miR-142, miR-149-3p, miR-151, miR-16-1, miR-1826, miR-183-3p, miR-21,miR-204-5p, miR-218, miR-219, miR-26a-1, miR-292, miR-299-5p, miR-30, miR-300, miR-335,miR-345, miR-345, miR-374b, miR-375,miR-451a, miR-486-5p, miR-514a-3p, miR-613, miR-637, miR-662, miR-873, miR-876-3p, miR-939 | mRNA cleavage or translation blockage | LOC10050766 | NONHSAT037832 | 39–46,56,59,61,64,73–95 |
| PVT11, BANCR | |||||||||||
| FTC | ZIC1, KISS1R, RASSF1, PTEN | Metastasis, cellular proliferation | Acetylation | Apoptosis reduction and inhibition cell cycle arrest | miR-125a-3p, miR-142-3p, miR-146, miR-155, miR-181, miR-182, miR-183, miR-187, miR-197, miR-200, miR-21, miR-221, miR-222, miR-224, miR-346, miR-597, miR-96 | let-7, miR-142-3p, miR-1247, miR-144, miR-150,,miR-191, miR-192, miR-197, miR-199a-5p,miR-328 and miR-346, miR-455, miR-542-5p, miR-574-3p | mRNA cleavage or translation blockage | PVT11 | 39–46,62,63,67,96–100 | ||
| ATC | NOTCH4, TCL1B | Metastasis, cellular proliferation | Acetylation | Apoptosis reduction and inhibition cell cycle arrest | miR-17-92, miR-146, miR-18a, miR-20a, miR-21, miR-221, miR-222, miR-92 | let-7, miR-1, miR-125b, miR-138, miR-200, miR-25, miR-26a, miR-30a, miR-30d, miR-99a | mRNA cleavage or translation blockage | PVT11 | 42,60,63,64,67 | ||
| 101–109 | |||||||||||
| MTC | INSL4, DPPA2 | Metastasis, cellular proliferation | miR-10a, miR-21, miR-124a, miR-127,miR-129, miR-137, miR-154, miR-183, miR-21, miR-224, miR-323, miR-370, miR-375, miR-9 | miR-9-3p, miR-129-5p, miR-455 | mRNA cleavage or translation blockage | 45,110–115 | |||||
ATC: anaplastic thyroid cancer; FTC: follicular thyroid cancer; PTC: papillary thyroid cancer; PDTC: poorly differentiated thyroid carcinoma.
In PTC cell lines, overexpression of miR-146b-5p is associated with greater invasion of tumoral cells.73 This role in malignancy and extra-thyroid invasiveness is due to the targeting of SMAD4, a protein of the SMAD family involved in the signal transduction of TGF-β (tumor growth factor-β) which has a role in cell growth, differentiation, apoptosis and cell motility.74,75 In addition, Czajka et al.76 confirmed the role of miR-146b-5p in PTC aggressiveness as being due to the down-regulation of RARβ (retinoic acid receptor beta) protein, a frequent event in PTC, the lack of RARβ contributing to the ineffectiveness of retinoic acid and radioactive iodine treatment. The up-regulation of miR-146b-5p observed exclusively in PTCs and FVPTCs has converted this miRNA into a useful biomarker for the PTC subtype.77
Increased levels of the miRNA 222 and miRNA 221 families have also been associated with aggressive PTC variants. Both are involved in cell proliferation and cell cycle and apoptotic processes, as well as invasion, metastasis and angiogenesis.78 Their role in aggressive forms was confirmed by Cong et al.79 and by Yoruker et al.,80 both studies demonstrated the down-regulation of these miRNAs after tumor resection in PTC patients, providing support for the possibility of using these molecules as biomarkers of PTC recurrence. Recently, Rossi et al.81 have demonstrated the role of miR-375 as a positive biomarker in the identification of the malignant form of follicular neoplasm in fine needle aspiration cytology, and it has potential to be used in the criteria for deciding the most appropriate treatment. A wide range of miRNAs are up-regulated in PTCs, though not all of them have a potential role as biomarkers in diagnosis or prognosis in thyroid cancer. The details of these differences have been summarized by Zhang et al.82 and we describe them in Table 2.
Focusing on down-regulated miRNAs, Minna et al.83 identified miR-451 reduction in aggressive forms of PTC, something which would exert an antitumor function by decreasing AKT/mTOR signaling or miR-137.84 Another miRNA with a tumor suppressor role and reduced levels in tumor samples is miR-375. It interacts with the ERBB2 (Her2/neu) gene which codifies for a tyrosin quinase involved in EGFR signaling, inhibiting cell proliferation.85 Analogous results were obtained with miR-31.86,87 A potential biomarker role has also been assigned to miR-7-5p and miR-204-5p. Reduced levels of these miRNAs have been found to be significantly associated with BRAFV600E positive tumors.88 Their role as “aggressive markers” of PTC subtypes has also been confirmed by Saiselet et al.89 (Table 2).
Long non-coding RNAs (lncRNAs) have a regulatory role at transcriptional or post-transcriptional level, acting as a signal in response to a stimulus, and controlling processes such as cell cycle, cell differentiation and cell death.90 Lan et al.91 have developed the first genome-wide profile of lncRNAs in PTC. These authors found significant differences in the lncRNAs expressed between tumor/normal tissue pairs that were implicated in the regulation of genes involved in PTC development. Kim et al.92 have identified elevated levels of LOC100507661, in association with BRAFV600E mutation and lymph node metastasis, highlighting this lncRNA as a potential biomarker, or as a target in the aggressive forms of PTC. PVT1 is other lncRNA with elevated levels in thyroid cancer cell lines, as well as in thyroid samples.93 Up-regulation of BRAF-activated long non-coding RNA (BANCR) or NONHSAT037832 down-regulation are also now also considered biomarkers of PTC.94,95
Follicular thyroid carcinomaThe differential values of miRNAs found inFTCs are also frequently present in PTCs, FAs and FVPTCs. This feature makes it difficult to find an miRNA which can be used as an exclusive marker for this thyroid tumor subtype (Table 2).
Up-regulation of miR-221 in FTCs compared with a normal paired sample was described by Wojtas et al.,96 but this increment was not associated with extra-thyroid invasion and lymph node metastasis, as was observed in the PTC variants. These authors suggest the possibility that miR-221 increment in FTC is an early event in the transformation of follicular cells, and that it is enhanced during malignant transformation. Increases in miR-146b in the FTC variant, when compared with paired unaffected tissue, was also observed by the same authors,96 calling into question the idea that miR-146b is an exclusive marker of PTC variants and its use as a biomarker for the differentiated thyroid tumors.
miR-191 levels are reduced in FTC samples compared with normal samples, and this event has also been observed in FAs, supporting the idea that it is an initial event in tumoral development.97 The protein CDK6, a cyclin, seems to be the specific target of this miRNA. CDK6 has pro-oncogenic properties, which is why its increased expression due to the reduction in miR-191 leads to follicular thyroid neoplasia.
miR-142-3p is also described as having a tumor suppressor role in these tumors.98 Low levels of this miRNA were identified in FTCs compared with non-tumoral tissues. ASH1L (Absent Small and Homeotic Disks Protein 1 Homolog) and MLL1 (Mixed-Lineage Leukemia) are the targets of this miRNA and they act as activating transcription factors for the HOX gene and others, such as metalloproteases, and angiogenic factors that contribute to cancer development. Down-regulation of miR-199a-5p in a population of FTC samples was observed by Sun et al.99 This miRNA targets the CTGF (connective tissue growth factor) gene. CTGF protein seems to have a role in proliferation, differentiation and cell adhesion in many cell types, as well as in tumor development,100 providing evidence to support its potential role in FTC development and its use as a biomarker for this disease.
With respect to lncRNAs, only PVT1 demonstrated elevated levels in thyroid cancer cell lines as well as in thyroid samples,93 as has been previously indicated in the section on PTC.
Anaplastic thyroid carcinomaAs already mentioned, the role of miRNAs in undifferentiated thyroid tumors has been described (view summary in Table 2). Anaplastic thyroid tumors are very aggressive and frequently associated with being unresponsive to therapies.
miR-17-92 has been reported to have oncogenic properties in ATC tumors and to be over-expressed in relation to normal tissues.101 This miRNA seems to develop its oncogenic role by regulating levels of PTEN, which acts as a negative regulator of the PIK3 signaling pathway.102 Shao et al.103 demonstrated that miR-4295 expression was able to promote proliferation and invasion in ATC cell lines. The aggressive behavior observed was due to a reduction in the CDKN1A (cyclin-dependent kinase inhibitor 1A) gene, the target of miR-4295. CDKN1A, also known as p21, is involved in cell growth regulation. It prevents cell cycle progression in the G1 phase by blocking cyclin-CDK2/CDK4 complexes.104
Other miRNAs with an elevated presence in ATCs are miR-146, miR-221 and miR-222. They all seem to be involved in tumor size, metastasis and recurrence, through their interaction with the NF-kB signaling pathway, or through the regulation of target genes such as CDKN1B, which controls cell cycle, or RECK, a metalloprotease inhibitor.81 In addition, Haghpanah et al.105 have identified miR-21 as a potential oncogenic miRNA in ATCs.
Similar to the FTCs, reductions in miRNA levels that appear to be related to the aggressive forms have been observed in ATCs. The miR-200, miR-30 and let-7 families are some examples. These miRNA participate in the TGF-beta or EGFR (epidermal growth factor receptor) signaling pathways and in the control of the protein expression that regulates tumoral cell development.106
miR-138 levels have been found decreased in ATCs.107 This reduction is associated with increased expression of the hTERT gene, which contributes to tumor development. miR-30a down-regulation has also been described by Boufraqech et al.,108 who ascribed ita tumor suppressor role in cell lines and in murine models. Similarly, miR-30d and miR-99a are also down-regulated in ATCs.109
Medullary thyroid carcinomaMTCs represent 5–10% of thyroid cancers. While 75% of these are associated with sporadic forms, 25% are hereditary and are due to mutations on the RET-oncogene. miRNAs are also associated with this subtype (Table 2). Puppin et al. demonstrated an association between the genes involved in miRNA formation, like XPO5 (Exportin 5), DICER, DROSHA and DGCR8, and mutated RET forms of MTC.110
A group of miRNAs – 127, miR-154, miR-224, miR-323, miR-370, miR-183, miR-375, and miR-9 – were found raised in MTCs. Some of these miRNAs are involved in the inhibition of tumor suppressor genes, thus contributing to the aggressive phenotype.111 Further new miRNAs have more recently been acknowledged to be related to MTC development. One of them, miR-21, is frequently found elevated in this type of tumor and is associated with permanent disease.112PDCD4, a tumor suppressor gene, is the target of miR-21, and has been found down-regulated in thyroid disease, especially in the aggressive forms, and this was associated with an increase in miR-21. The target gene exerts its antitumor function by controlling the translation of proteins that allow cells to avoid apoptosis; hence its reduction contributes to cancer progression.
Two other miRNAs, miR-375 and miR-10a, are also elevated in MTC. miR-375 targets the YAP1 protein which acts as a transcriptional co-activator of genes with oncogenic or suppressor actions.113
In contrast, miR-129-5p is found diminished in MTCs. This miRNA develops its antitumor functions by decreasing the AKT pathway, which leads to an increment in cellular apoptosis, and by blocking cell migration.114 It does this by reducing AKT phosphorylation levels, but it can also bind to the 3′-UTR of the RET gene, responsible for the hereditary forms of MTC, and thus blocks its expression. miR-9-3p is also down-regulated in MTC cell lines.115 The gene target of this miRNA is the Beclin 1 gene, and, as was described with respect to ATC, it is involved in autophagy processes and its down-regulation can increase the sensitivity of tumors to chemotherapeutic drugs.
ConclusionMethodologies for the analysis of epigenetic modifications have allowed the development of genome-wide profiles and the identification of the epigenome of different tissues, both in healthy and pathological conditions. In this review, we have focused on the epigenetic modifications associated with the different thyroid cancer subtypes compared with paired non-tumoral samples (Table 2).
We have described here the differences found in the epigenomes of healthy and diseased tissue and their roles in thyroid cancer development, progression and response to treatment. As more genome-wide epigenome data becomes accessible we will be able to better understand the interaction between various epigenetic modifications and their role in gene regulation and chromatin structure. Among these changes, DNA methylation and miRNA seem to have the greatest importance in tumor prognosis, suggesting the possibility of developing promising new therapies to treat thyroid tumors, especially the more aggressive forms, which are focused on demethylating agents, histone deacetylase inhibitors, and the development of mature miRNAs to mimic or block gene expression.
Conflict of interestsThe authors declare that they have no conflict of interest.
This work was supported by grants from the Fundación Mutua Madrileña (IX Convocatoria de Ayudas para la Investigación Clínica-2012), Instituto de Salud Carlos III (FIS PI11/02795 and FIS P14/00970) and the Sociedad Española de Diabetes (SED) (in 2012). Special thanks go to Ronnie Lendrum, the English style editor, for her critical, constructive reading and invaluable comments.
