





Antibodies against a protein complex that includes voltage-gated potassium channels (VGKC) have been reported in patients with limbic encephalitis, peripheral nerve hyperexcitability, Morvan's syndrome, and a large variety of neurological syndromes.
Review summaryIn this article, a review is presented of the syndromes associated with antibodies against VGKC-related proteins and the main antigens of this protein complex, the proteins LGI1 (leucine rich glioma inactivated protein 1) and Caspr2 (contactin-associated protein-like 2). The conceptual problems and clinical implications of the description of antibodies against VGKC-related proteins other than LGI1 and Caspr2 are also discussed.
Although initial studies indicated the occurrence of antibodies against VGKC, recent investigations have shown that the main antigens are a neuronal secreted protein known as LGI1 which modulates synaptic excitability, and a protein called Caspr2 located on the cell surface and processes of neurons of different brain regions, and at the juxtaparanodal region of myelinated axons. While antibodies against LGI1 preferentially associate with classical limbic encephalitis, antibodies against Caspr2 associate with a wider spectrum of symptoms, including Morvan's syndrome, peripheral nerve hyperexcitability or neuromyotonia, and limbic or more extensive encephalitis. In addition there are reports of patients with antibodies against VGKC-related proteins that are different from LGI1 or Caspr2. In these cases, the identity and location of the antigens are unknown, the syndrome association is not specific, and the response to treatment uncertain.
ConclusionsThe discovery of antigens such as LGI1 and Caspr2 has resulted in a clinical and molecular definition of the broad group of diseases previously attributed to antibodies against VGKC. Considering the literature that describes the presence of antibodies against VGKC other than LGI1 and Caspr2 proteins, we propose a practical algorithm for the diagnosis and treatment of these patients.
Los anticuerpos contra un complejo proteico que incluye a los canales de potasio dependientes de voltaje (CKVD) se han descrito en pacientes con encefalitis límbica, hiperexcitabilidad del nervio periférico, síndrome de Morvan, así como en un creciente grupo de síndromes neurológicos.
DesarrolloEn este artículo revisamos los síndromes asociados a anticuerpos contra proteínas relacionadas con los CKVD y los 2 antígenos principales de este complejo, las proteínas leucine rich glioma inactivated protein 1 (LGI1) y contactin-associated protein-like 2 (Caspr2). Así mismo describimos los problemas conceptuales y las implicaciones diagnósticas de la descripción de anticuerpos contra CKVD diferentes de LGI1 y Caspr2.
Aunque inicialmente se consideró que existían anticuerpos dirigidos contra CKVD, recientemente se ha identificado que, en la mayor parte de los casos, los antígenos son una proteína neuronal secretada denominada LGI1, involucrada en el control de la excitabilidad sináptica, y la proteína Caspr2, localizada en la superficie neuronal de varias regiones cerebrales y en la región yuxtaparanodal de axones mielinizados. Mientras que los anticuerpos contra LGI1 se asocian preferentemente a un cuadro clásico de encefalitis límbica, los anticuerpos contra Caspr2 muestran un espectro clínico más amplio, incluyendo el síndrome de Morvan, la hiperexcitabilidad del nervio periférico o neuromiotonía, o una encefalitis límbica o difusa. Existen además casos descritos de pacientes con anticuerpos contra el complejo CKVD que no tienen anticuerpos contra LGI1 o Caspr2. En estos casos, la identidad y la localización de los antígenos es desconocida, la asociación sindrómica inespecífica y la respuesta al tratamiento, incierta.
ConclusionesEl descubrimiento de los antígenos LG1 y Caspr2 ha permitido delimitar clínica y molecularmente el amplio grupo de síndromes previamente atribuidos a anticuerpos contra CKVD. Frente a la literatura que describe la presencia de anticuerpos contra CKVD diferentes a LGI1 y Caspr2, proponemos un algoritmo práctico para el diagnóstico y el tratamiento de estos pacientes.
Voltage-gated potassium channel (VGKC) antibodies have been identified in a wide range of neurological syndromes involving the central and peripheral nervous systems in both adults1 and children.2 These antibodies were initially thought to target epitopes of the VGKC; however, research in the past few years indicates that most of them are directed to leucine-rich glioma inactivated protein 1 (LGI1)3 and contactin-associated protein-like 2 (CASPR2).3,4 Furthermore, recent studies have described a group of patients testing positive for antibodies against VGKC-complex proteins but negative for Caspr2 and LGI1.5,6
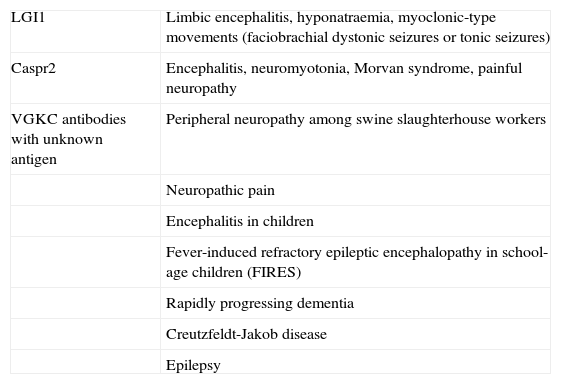
Anti-LGI1 antibodies are present in limbic encephalitis,3 while anti-Caspr2 antibodies may be associated with encephalitis,3,4 peripheral nerve hyperexcitability (also known as acquired neuromyotonia or Isaacs syndrome),7 or a combination of both (Morvan syndrome).3–6 These 2 proteins are well characterised, and alterations in them provide the pathophysiological mechanism for the clinical symptoms of each type of autoimmune response. In contrast, target antigens in patients with antibodies against VGKC-complex proteins, but testing negative for LGI1 and Caspr2, are unknown.8 Patients with these antibodies form a heterogeneous and increasing population. For the above reasons, current research focuses on determining the clinical significance and pathogenic mechanisms of these antibodies (Table 1). The present review article aims to clarify these questions related to the clinical and pathological spectra and describe the syndromes associated with these antibodies.
Clinical spectrum associated with presence of VGKC antibodies
| LGI1 | Limbic encephalitis, hyponatraemia, myoclonic-type movements (faciobrachial dystonic seizures or tonic seizures) |
| Caspr2 | Encephalitis, neuromyotonia, Morvan syndrome, painful neuropathy |
| VGKC antibodies with unknown antigen | Peripheral neuropathy among swine slaughterhouse workers |
| Neuropathic pain | |
| Encephalitis in children | |
| Fever-induced refractory epileptic encephalopathy in school-age children (FIRES) | |
| Rapidly progressing dementia | |
| Creutzfeldt-Jakob disease | |
| Epilepsy |
The term ‘VGKC antibody’ has been used to denote antibodies detected with radioimmunoassay (RIA) that labels the protein complex including the Kv1.1 and Kv1.2 subunits of the Shaker family of VGKCs. This test yields positive results when the radiotracer 125I-α-dendrotoxin binds to antigens of a protein complex which precipitates along with VGKC proteins. However, this technique cannot be used to identify the antigen, or even its type (neuronal, axonal, or synaptic).
Meanwhile, immunofluorescence tests in hippocampal neuron cultures have shown that antigens attributed to VGKC are located on the surface of neurons. These antigens have been identified by precipitating them with patients’ antibodies and sequencing them. Once they had been identified as LGI13 or Caspr2,4 researchers developed highly specific diagnostic techniques using cells that express these antigens, such as cell-based assay (CBA) (Fig. 1).
 and a patient with antibodies against Caspr2 (D and E) shows an intense reactivity with neuropil. This is not observed with CSF from a control patient (G and H). Immunocytochemistry demonstrates the neuronal surface location of the antigens (LGI1 and Caspr2) with rat hippocampal cell cultures incubated with CSF samples from the same patients (C and F). No reactivity between CSF and neuronal cultures is observed in the control patient (I). Antigen identities (LGI1 and Caspr2) were demonstrated in the transfected HEK293 cells (CBA) expressing these proteins (not shown).")
CSF reactivity in patients with antibodies against LGI1 or Caspr2 in rat brain and neuronal cultures. Immunohistochemistry in rat brain using CSF from a patient with antibodies against LGI1 (A and B) and a patient with antibodies against Caspr2 (D and E) shows an intense reactivity with neuropil. This is not observed with CSF from a control patient (G and H). Immunocytochemistry demonstrates the neuronal surface location of the antigens (LGI1 and Caspr2) with rat hippocampal cell cultures incubated with CSF samples from the same patients (C and F). No reactivity between CSF and neuronal cultures is observed in the control patient (I). Antigen identities (LGI1 and Caspr2) were demonstrated in the transfected HEK293 cells (CBA) expressing these proteins (not shown).
Despite these advances, some researchers support using RIA to detect VGKC-complex antibodies, and they have reported that these antibodies are detected in patients negative for LGI1 and Caspr2 in 39% to 68% of all cases.5,6 The question raised by these studies, which do not use supplementary techniques to specify the antigen type, will be addressed in a later section (‘VGKC antibodies other than LGI1 and Caspr2’).
LGI1 is a neuronal secreted protein that interacts with presynaptic ADAM23 and postsynaptic ADAM22 to organise a transsynaptic protein complex that has been linked to epilepsy in humans.9 Other components of this complex include presynaptic Kv1.1 and Kv1.2 subunits and postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. Mutations in the gene that codes for LGI1 have been associated with autosomal dominant lateral temporal lobe epilepsy and autosomal dominant partial epilepsy with auditory features,10,11 the latter being a type of hereditary epilepsy characterised by partial seizures and visual or auditory hallucinations. This mutation in animal models has been correlated to increased neuronal excitability, which in turn has been linked to decreased AMPA receptor activity in inhibitory neurons9 and increased glutamate release.12 Some suggest that this hyperexcitability contributes to the memory impairments and epilepsy exhibited by patients with anti-LGI1 antibodies.3
Caspr2 is an axonal transmembrane protein of the neurexin superfamily that binds to contactin-2. It is involved in the clustering of Kv1 potassium channels in the juxtaparanodal region, where it seems to promote the normal function of myelinated axons.13 Caspr2 is also present in the hippocampus and cerebellum.14 Mutations and polymorphisms of the gene that codes for Caspr2 (CNTNAP2) have been detected in psychiatric patients, in those with drug-resistant epilepsy, and in cases of peripheral nerve hyperexcitability.14–17 These findings support the pathophysiological basis of clinical symptoms associated with presence of anti-Caspr2 antibodies.
Clinical spectrum associated with anti-LGI1 antibodiesAnti-LGI1 antibodies have been associated with symptoms of classic limbic encephalitis, which is characterised by short-term memory loss and epileptic seizures. From an epidemiological perspective, findings by Lai et al.3 show that it is more common in men in about the sixth decade, although it has been known to occur in ages from childhood to 80 years. Its typical clinical presentation is one of subacute limbic encephalitis, which may be associated with a variety of symptoms presenting in any order. This entity eventually elicits an amnestic deficit that is frequently associated with REM sleep behaviour disorder18 and epileptic seizures in a high percentage of cases. Epilepsy may manifest as generalised seizures, mesial temporal lobe seizures, and tonic seizures in up to 40% of all cases.19 The frequency of tonic seizures in this type of cases has led researchers to consider them a warning sign in the differential diagnosis of limbic encephalitis.19 They typically present as sudden brief spasmodic asymmetrical movements; they are predominantly tonic, although they can also be dystonic in the orofacial area and upper limbs. For this reason, some authors call them faciobrachial dystonic seizures.20 However, this term can be misleading for 3 reasons: (1) movements are not restricted to the upper limbs and orofacial region in all cases; (2) there is an electroencephalographic correlation between these seizures and decreased alpha frequency, which is characteristic but not pathognomonic of tonic seizures; and (3) some patients respond to antiepileptic treatment.20 The last 2 points suggest that the most probable underlying pathogenic mechanism is epileptogenic rather than one related to movement disorders.
Regarding complementary studies, associated hyponatraemia has been found in up to 60% of these patients.3,6 Results from routine cerebrospinal fluid (CSF) tests are usually normal, although patients may present moderate lymphocytosis and increased protein levels. In MRI T2-weighted sequences, 84% of the patients display a unilateral or bilateral increase in signal intensity in the medial temporal region.
Co-presence of tumours is rare, and most such cases are thymomas. In 70% to 80% of patients, symptoms respond to treatment with steroids, intravenous immunoglobulins, or plasma exchange. However, memory impairments persist in many patients, which makes it difficult for them to return to normal duties at work. Recurrence of encephalitis is also rare.
Clinical spectrum associated with anti-Caspr2 antibodiesAnti-Caspr2 antibodies are associated with symptoms affecting the central nervous system (encephalitis), the peripheral nervous system (peripheral nerve hyperexcitability), or both (Morvan syndrome). It is difficult to determine the frequency of presentation of these forms and whether one of them predominates over the rest, since only a few patients have been included in the series published to date.
Encephalitis associated with presence of anti-Caspr2 antibodies usually manifests around the age of 60, although it has been described in patients aged 19 to 80, predominantly in men.4,21 The typical clinical manifestation is diffuse, multifocal, or limbic encephalitis, which in most cases is associated with peripheral nerve hyperexcitability. It presents as neuromyotonia, or cramps and fasciculations in incomplete forms. Neuromyotonia may precede development of encephalitic symptoms for a variable period of time. Presence of epileptic seizures, including tonic seizures, is not infrequent.4,21
Patients with Morvan syndrome are described as having a higher prevalence of sleep disorders; insomnia is the most common feature,4,21 which may at times result in agrypnia excitata.22 Furthermore, up to 62% of the cases present neuropathic pain frequently associated with areflexia and hypoesthesia with a stocking-and-glove pattern. Autonomic dysfunction, in the form of hyperhidrosis or cardiovascular instability, may present in up to 93% of the cases.21 On the other hand, some patients have been found to exhibit bulbar symptoms, fasciculations, and lower limb weakness associated with presence of antibodies against acetylcholine receptors and anti-muscle-specific kinase antibodies. This has led to including motor neuron disease in the differential diagnosis.4
Results from the CSF analysis are abnormal in up to 25% of all patients. This generally manifests as either pleocytosis or elevated protein levels, plus oligoclonal bands in certain cases.4 MRI scans may also reveal increased signal activity in T2-weighted temporal medial lobe sequences,21 although this finding is normal in patients with Morvan syndrome.4,5,21
Regarding associated tumours, the data available in the literature vary. While some researchers have found tumours in 20% to 41% of patients5,21 (predominantly thymomas in those patients with Morvan syndrome), other studies report lower rates.4 Regarding treatment, prognosis seems to be good overall, except in the cases associated with tumours.4,5,21 However, relapses are not infrequent.
Although the presence of VGKC-complex antibodies has traditionally been associated with isolated acquired neuromyotonia,23,24 more recent studies suggest that this association is in fact less frequent than was previously believed.25
Syndromes and symptoms associated with antibodies against voltage-gated potassium channel complex: unknown antigens and uncertain clinical valueAlthough detection of LGI1 and Caspr2 antibodies has helped clarify the clinical spectrum of syndromes associated with VGKC antibodies, there is also a heterogeneous group of clinical syndromes linked to these VGKC antibodies in which LGI1 and Caspr2 antibodies are not present. This wide range of syndromes includes not only diseases restricted to the central nervous system (fever-induced refractory epileptic encephalopathy, epilepsy, encephalitis in children, rapidly progressing dementia, and Creutzfeldt-Jakob disease) but also those affecting the peripheral nervous system (neuropathy, neuropathic pain).2,26–29 The precise antigens are unknown in all these cases: it is not even clear whether they are intracellular or extracellular. Furthermore, responses to immunosuppressive treatment in the cases described above vary greatly, which highlights the relevance of the diagnostic value and pathogenesis of these antibodies. The low diagnostic value of these antibodies was demonstrated by a recent study in which authors classified syndromes as autoimmune or non-autoimmune depending on response to immunotherapy, regardless of presence or absence of VGKC antibodies.30 Further research is necessary to understand the clinical significance of VGKC antibodies.
Practical diagnostic and therapeutic management of patients with VGKC antibodiesIn light of the above, we propose using the algorithm shown in Fig. 2 when there is clinical suspicion of an autoimmune neurological syndrome associated with presence of VGKC-complex antibodies. The solid lines indicate the recommended steps. The dotted lines indicate that, regardless of the result from RIA, the study will require a diagnostic confirmation with more specific techniques, such as immunocytochemistry with cells expressing LGI1 and Caspr2. If this last test is negative, additional tests with neuronal cultures must be performed to determine whether or not antigens are located on the cell surface. Reactivity of serum and/or CSF with the surface of living neurons indicates a high probability that the syndrome will respond to immunotherapy, which must therefore be started regardless of the identity of the antigen. On the other hand, if serum and/or CSF do not react with the surface of living neurons but do react with sections of rat brains, antigens are intracellular or onconeuronal. In this case, analysing classic paraneoplastic antibodies is recommended; when results are positive, tumours should be localised and treated. Syndromes associated with antibodies against intracellular or paraneoplastic antigens tend to have a poorer response to immunotherapy than those associated with antibodies against neuronal surface antigens.

Diagnostic algorithm for patients with antibodies against VGKC-related proteins. *Clinical syndromes listed in Table 1. **NMDA receptor, AMPA receptor, GABAb receptor, DPPX, Gly receptor, others.
The most frequently used first-line immunotherapies for LGI1, Caspr2, and other surface antigens are steroids, intravenous immunoglobulins, and plasma exchange, or a combination of the above. On a practical level, the most frequent combination is steroids plus intravenous immunoglobulins. When patients do not respond to these immunotherapies, the treatment used in patients with anti-NMDA receptor encephalitis (rituximab and/or cyclophosphamide) is the most reasonable option.31
Conflict of interestDr Dalmau owns the patent for the use of Ma2 and NMDA receptor autoantibody diagnostic tests, and has filed a patent application for the use of GABAa and GABAb receptor autoantibody diagnostic tests.
FundingDr Dalmau is funded by the National Institutes of Health (NIH RO1NS077851), Fundació la Marató TV3, and Institute of Health Carlos III (FIS, PI11/01780).
Please cite this article as: Montojo MT, Petit-Pedrol M, Graus F, Dalmau J. Espectro clínico y valor diagnóstico de los anticuerpos contra el complejo proteico asociado a canales de potasio. Neurología. 2015;30:295–301.
Dr Montojo is a neurology resident at Hospital Universitario Fundación de Alcorcón, Madrid. The present study was performed while she was working at the immunology laboratory at IDIBAPS-Hospital Clínic, Barcelona.
articles

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals