




Las malformaciones pulmonares corresponden a distintas anomalías del sistema respiratorio que se presentan con baja incidencia (1 en cada 10.000 a 35.000 embarazos), dentro de las que se incluye a las Malformaciones Pulmonares Congénitas y de la vía aérea (MCPA), antes conocida malformación adenomatosa quística, secuestros pulmonares, lesiones híbridas y enfisema lobar congénito. Durante los últimos años se ha visto un aumento en el diagnóstico antenatal y avances en el conocimiento de la patogénesis e historia natural de esta enfermedad, pero aún existe controversia en cuanto a la clasificación a utilizar y a su tratamiento. La mayoría de los recién nacidos (90%) son asintomáticos al nacer, pero hay malformaciones que generan serias complicaciones para el feto o recién nacido. El propósito de esta publicación es hacer un resumen actualizado de la historia natural, diagnóstico y tratamiento de las MCPA.
Pulmonary malformations correspond to different abnormalities of the respiratory system that occur with low incidence (1 in every 10000 to 35000 pregnancies), within which are included Lung Congenital Malformations and airway (MCPA) formerly known malformation adenomatous cystic, pulmonary sequestrations, congenital lobar hybrid lesions and emphysema. In recent years there has been an increase in antenatal diagnosis and advances in the understanding of the pathogenesis and natural history of this disease, but there is still controversy regarding the classification to be used and its treatment. Most newborns (90%) are asymptomatic at birth, but there are defects that create serious complications for the fetus or newborn. The purpose of this publication is to make an updated summary of the natural history, diagnosis and treatment of MCPA.
Dentro del concepto de malformaciones pulmonares se ha incluido a un grupo diverso de anomalías que ocurren durante el proceso de formación del sistema respiratorio, que puede tener distintas manifestaciones clínicas y pronósticos. Son alteraciones poco frecuentes que se presentan con una incidencia aproximada de 1 en cada 10000 a 35000 embarazos.
A lo largo de los años han ocurrido cambios en su nomenclatura y clasificación a medida de que se han incorporado nuevos conocimientos en patología, pero aún no existe consenso acerca de la clasificación a utilizar. Dentro de este grupo de anomalías se incluye a las Malformaciones Congénitas Pulmonares y de la Vía Aérea (MCPA), previamente llamadas malformaciones adenomatoídea quísticas (CCAM o MAQ), a los quistes broncogénicos, secuestros pulmonares, atresias bronquiales, la hiperinsuflación lobar congénita y a lesiones híbridas. En este artículo nos enfocaremos en las MCPA, que son las que se presentan con mayor frecuencia (95% de los casos), con una incidencia estimada en 3,5 por 10.000 RNV1,2.
El diagnóstico precoz de las MCPA ha aumentado por el uso rutinario de la ecografía antenatal y por la mayor disponibilidad de Resonancia Nuclear Magnética (RM) en etapa fetal. La mayoría de los recién nacidos (90%) son asintomáticos al nacer, existiendo un grupo de malformaciones que involucionan en la etapa final del embarazo, mientras que otras aumentan de tamaño, generando serias complicaciones para el feto y/o recién nacido. Además del progreso en el diagnóstico de las MCPA, han ocurrido avances en el manejo pre y post natal, sobre todo con a la introducción de la cirugía fetal y la incorporación de nuevas técnicas quirúrgicas, pero aún existen controversias en el manejo de los pacientes con MCPA.
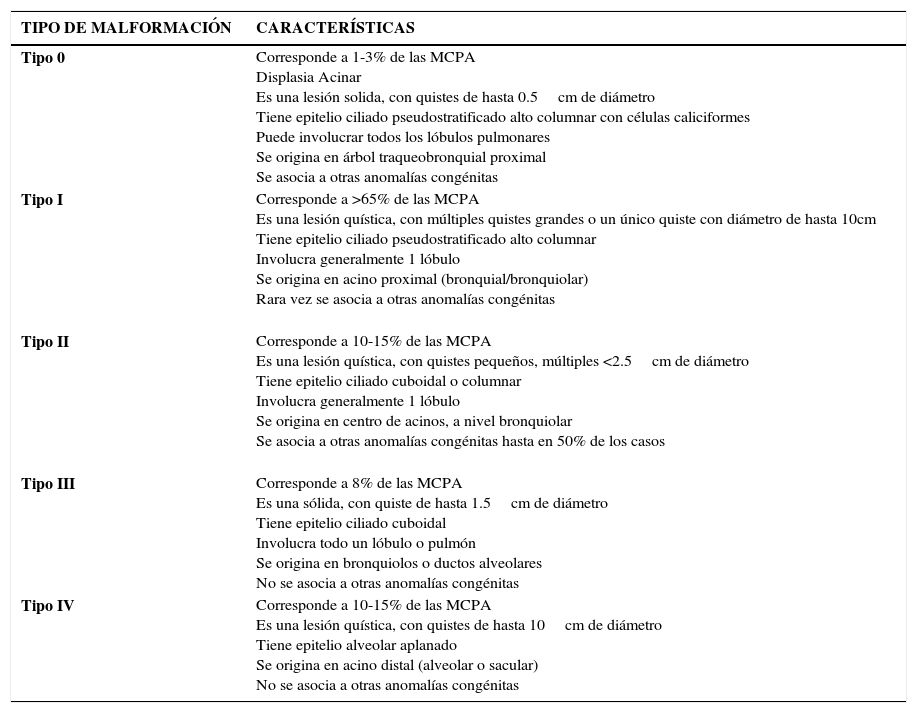
ClasificaciónLa clasificación de las malformaciones pulmonares ha sufrido variaciones desde que Stocker et al. en 1977 clasificara las MAQ en tres tipos histológicos; 1, 2 y 3 en base a su evolución clínica y al aspecto macro y microscópico5–7. En el año 2008 se amplió esta clasificación incluyendo los tipos 0 y IV y el mismo Stocker propone cambiar el nombre de CCAM a MCPA, ya que las malformaciones involucran diferentes partes del árbol traqueobronquial y las lesiones quísticas se encuentran sólo en tres de los cinco tipos descritos8. En la Tabla 1 se muestran las principales características de cada tipo de MCPA.
CLASIFICACIÓN DE MALFORMACIONES CONGÉNITAS PULMONARES Y DE LA VÍA AÉREA (MCPA)
| TIPO DE MALFORMACIÓN | CARACTERÍSTICAS |
|---|---|
| Tipo 0 | Corresponde a 1-3% de las MCPA Displasia Acinar Es una lesión solida, con quistes de hasta 0.5cm de diámetro Tiene epitelio ciliado pseudostratificado alto columnar con células caliciformes Puede involucrar todos los lóbulos pulmonares Se origina en árbol traqueobronquial proximal Se asocia a otras anomalías congénitas |
| Tipo I | Corresponde a >65% de las MCPA Es una lesión quística, con múltiples quistes grandes o un único quiste con diámetro de hasta 10cm Tiene epitelio ciliado pseudostratificado alto columnar Involucra generalmente 1 lóbulo Se origina en acino proximal (bronquial/bronquiolar) Rara vez se asocia a otras anomalías congénitas |
| Tipo II | Corresponde a 10-15% de las MCPA Es una lesión quística, con quistes pequeños, múltiples <2.5cm de diámetro Tiene epitelio ciliado cuboidal o columnar Involucra generalmente 1 lóbulo Se origina en centro de acinos, a nivel bronquiolar Se asocia a otras anomalías congénitas hasta en 50% de los casos |
| Tipo III | Corresponde a 8% de las MCPA Es una sólida, con quiste de hasta 1.5cm de diámetro Tiene epitelio ciliado cuboidal Involucra todo un lóbulo o pulmón Se origina en bronquiolos o ductos alveolares No se asocia a otras anomalías congénitas |
| Tipo IV | Corresponde a 10-15% de las MCPA Es una lesión quística, con quistes de hasta 10cm de diámetro Tiene epitelio alveolar aplanado Se origina en acino distal (alveolar o sacular) No se asocia a otras anomalías congénitas |
Otra clasificación bastante utilizada es la de Langston5, que simplifica el problema dividiendo a las lesiones quísticas es dos: (i) quistes grandes, equivalente a la tipo1 de Stocker y (ii) quistes pequeños, equivalente a la tipo 2 de Stocker, similar a lo planteado en la clasificación de Adzick et al.6, pero que en este caso está basada en la apariencia ecográfica de las lesiones y las diferencia en macroquísticas (>5mm de diámetro mayor) o microquísticas (<5mm) y que ha mostrado ser útil por asociarse a elementos pronósticos. Las macroquísticas tienden a crecer más lentamente y asociarse a un mejor pronóstico, mientras que las microquísticas, que tienden a crecer más rápido y se asocia con mayor frecuencia a desviación del mediastino, hipoplasia pulmonar, polihidroamnios e hidrops fetal, por lo que tendrán peor pronóstico6–8.
Los avances en conocimiento en cuanto al comportamiento y sobreposición de algunas malformaciones influenció la aparición de una nueva clasificación desarrollada por Langston, en que se incorpora además de las MCPA, otras malformaciones del sistema respiratorio y al blastoma pleuropulmonar, que es la que se muestra en la Tabla 29.
CLASIFICACIÓN DE MALFORMACIONES PULMONARES CONGÉNITAS
| Quiste Broncogénico |
| Atresia Bronquial • Aislada • Con conexión sistémica (secuestro intralobar) • Con conexión al tracto gastrointestinal • Conexión arterial sistémica en pulmón normal |
| CCAM, quistes grandes tipo I (tipo 1 de Stocker) • Aislada • Con conexión sistémica (híbrida, secuestro intralobar) |
| CCAM, quistes pequeños tipo II (tipo 2 de Stocker) • Aislada • Con conexión sistémica (híbrida, secuestro intralobar) |
| Secuestro extralobar • Con conexión al tracto gastrointestinal • Sin conexión al tracto gastrointestinal |
| Hiperplasia pulmonar y lesiones relacionadas • Atresia laríngea • CCAM sólida o adenomatoídea (tipo 3 de Stocker) • Lóbulo polialveolar |
| Hiperinsuflación lobar congénita |
| Otras lesiones quísticas • Quistes linfáticos, quistes digestivos, quistes mesoteliales, quistes simples pulmonares, blastoma pleuropulmonar tipo 1 |
(Ref. 9).
Las variaciones en la nomenclatura y la multiplicidad de clasificaciones ha impedido realizar un adecuado registro de las malformaciones pulmonares, establecer pronósticos y evaluar la efectividad de distintas conductas diagnósticas y terapéuticas. Es imprescindible contar con una clasificación común para mejorar estos aspectos.
Historia NaturalLa historia natural de las malformaciones pulmonares es difícil de predecir ya que algunas aumentan de tamaño durante el segundo trimestre de la gestación y pueden generar serias complicaciones para el feto o recién nacido, e incluso llevarlo a la muerte, mientras que otro grupo significativo de malformaciones involucionan en la etapa final del embarazo4. En la mayoría de los casos los fetos no presentan problemas antenatales resultando recién nacidos asintomáticos, describiéndose una sobrevida >95% en pacientes con lesiones quísticas y secuestros pulmonares6,10. Es posible ver una regresión de las lesiones previo al nacimiento hasta en un 68% de los secuestros y un 15% de las MCPA. La regresión completa se ha descrito, pero es muy infrecuente11.
Cuando las malformaciones pulmonares se asocian a hidrops fetal, lo que ocurre en un 5 a 30% de los casos11,12 el pronóstico empeora y se asocia a mayor mortalidad fetal6. En la revisión realizada por Witlox13 se observó una sobrevida postnatal de 92% en pacientes con secuestro y derrame, de 87% en MAQ sin hidrops y de 69% en MAQ con hidrops. Otros factores de mal pronóstico son el mayor tamaño de la lesión, el menor grado de desarrollo del pulmón no afectado y la presencia de otras malformaciones12. En la búsqueda de elementos pronósticos, el índice ecográfico CVR, que relaciona el volumen de la malformación con la circunferencia craneana del feto, normalizada a la edad gestacional, se ha validado como un buen índice pronóstico. Un índice CVR>1.6 se ha asociado a mayor riesgo de hidrops y a una peor evolución14. Este índice también sería útil para determinar la necesidad de programar un parto anticipado en centros de mayor complejidad, pero aún está en estudio15.
Histopatología y genéticaMariotti y cols. establecieron que el origen de las malformaciones pulmonares está en la alteración de la dicotomización del árbol respiratorio15–17 y el fenómeno inicial, según lo planteado por Langston, sería la obstrucción de la vía aérea9. Se ha mostrado que existe un aumento en la proliferación celular, asociado a una disminución de la apoptosis en estas malformaciones18 y se ha involucrado a distintos factores de crecimiento, que tendrían un patrón de expresión diferente en los pacientes. Liechty et al.19 mostraron que en las MCPA de crecimiento rápido, asociadas a hidrops, tenían elevado el factor BB derivado de plaquetas (PDGF-BB) en forma persistente. Otros factores involucrados serían el factor de transcripción I tiroideo (TTF1)17, factor neutrotrófico derivado de células gliales20 y los factores FGF9, FGF7 y FABP-721. También se ha detectado un aumento de expresión del gen Hoxb-522 y del factor de transcripción SOX2 que tendrían un rol crítico en la dicotomización del árbol respiratorio23. Otros autores han observado que la sobreexpresión del factor FGF10 en diferentes zonas y etapas del desarrollo pulmonar, estimularía la aparición de lesiones quísticas24.
DiagnósticoEl diagnóstico en el periodo antenatal se realiza fundamentalmente por medio de ecografía y cuando este examen otorga una información insuficiente debe complementarse con una RNM fetal. En la etapa antenatal no se puede establecer un diagnóstico definitivo por la frecuente sobre posición de malformaciones y por la evolución impredecible que éstas pueden tener. En esta etapa, las malformaciones deben describirse como macro, microquística o como lesión mixta y la caracterización debe incluir el tamaño y volumen de la malformación, su localización, la presencia o no de irrigación sistémica, de desviación de mediastino, derrame pleural u otros elementos de hidrops fetal. Después del nacimiento se debe hacer un seguimiento y estudio a todo paciente en que se haya sospechado una malformación pulmonar, el que debiera completarse antes de los 3 meses de vida, descartándose la presencia de otras malformaciones congénitas. La radiografía de tórax permitirá una aproximación inicial y es en muchos casos el examen por el cual se sospecha una malformación pulmonar en etapa postnatal, pero la tomografía computada (TC) es el examen de elección para confirmar el diagnóstico27 y asociada a angiografía será posible hacer un diagnóstico diferencial con mayor precisión l26. La RNM ha mostrado una capacidad más limitada que la TC para caracterizar adecuadamente las lesiones pulmonares28. Se debe considerar que el diagnóstico definitivo solamente se podrá establecer con la confirmación histológica.
TRATAMIENTO PRENATAL1. Uso de corticoides sistémicos antenatalExiste escasa evidencia que avale la efectividad del uso de corticoides en período antenatal. Los corticoides reducirían la producción de líquido dentro de la lesión y aumentarían su reabsorción, lo que ha sido observado en malformaciones microquísticas, pero no está claro su efecto en lesiones macroquísticas29. Algunos estudios han mostrado que el uso betametasona en el segundo trimestre del embarazo habría generado una disminución del tamaño de la malformación y reversión del hidrops y en otras series de casos se habría asociado a una disminución del índice CVR y una mejor sobrevida30,31. A pesar de que estos trabajos muestran beneficio del uso de corticoides, la evidencia actual no permite avalar su uso rutinario en el manejo de MCPA en el período antenatal.
2. Cirugía fetalLa cirugía fetal en pacientes con malformaciones pulmonares es una herramienta quirúrgica a considerar en pacientes con mal pronóstico in útero. Esta cirugía se puede realizar tanto durante el embarazo, mediante una laparotomía e histerotomía para llegar al feto y con una toracotomía proceder a una lobectomía. Posteriormente cerrar la toracotomía fetal, la Histerotomia y Laparotomia materna. También se puede realizar esta cirugía en EXIT (Ex Utero Intrapartum Therapy), durante una cesárea.
Las indicaciones para la cirugía fetal son:
- 1.
Malformaciones tipo sólida (micro-quísticas).
- 2.
Hidrops no respondedor a corticoides.
- 3.
Disfunción cardíaca.
- 4.
Sin otras alteraciones no asociadas a la malformación pulmonar.
- 5.
Cariotipo normal.
- 6.
Edad Gestacional <32 semanas.
Los recién nacidos con Malformaciones Congénitas Pulmonares en la gran mayoría de los casos son asintomáticos al nacer (90%) y los pacientes sintomáticos en general poseen complicaciones o signos prenatales que deben hacer prever un manejo pre y post natal adecuado32,33,42.
Es por esto que para enfrentar adecuadamente el manejo de los pacientes con MCPA deben dividirse en dos grupos:
- 1)
Pacientes Sintomáticos
- 2)
Pacientes Asintomáticos
Actualmente los avances en los cuidados intensivos neonatales han permitido estabilizar a los pacientes con malformaciones pulmonares que requieren de algún soporte al nacer debido a su malformación pulmonar (Insuficiencia respiratoria y/o cardiaca), requiriendo distintos métodos de apoyo desde el punto de vista hemodinámico y ventilatorio, pasando por el aporte adicional de oxígeno, la ventilación mecánica convencional y oscilatoria de alta frecuencia, el óxido nítrico e incluso ECMO (oxigenación por membrana extracorpórea) en caso de presentar hipertensión pulmonar persistente. Estos pacientes deben ser estabilizados antes de completar el estudio de su malformación, que incluye una evaluación cardiológica y estudio de imágenes torácicas con Angio-TC para tener la máxima información posible del punto de vista anatómico y funcional.
La gran mayoría de los pacientes deberá ser operados una vez estabilizados del punto de vista cardiopulmonar con la finalidad de resecar la malformación pulmonar ya sea con una lobectomía o segmentectomía.35,43,44. La cirugía podrá ser realizada mediante una toracotomía o con cirugía mínimamente invasiva (toracoscopía) dependiendo de las condiciones del paciente y la experiencia del equipo quirúrgico.
2) PACIENTES ASINTOMÁTICOSEl manejo de los recién nacidos con malformaciones pulmonares asintomáticos aún produce controversias. La decisión de realizar o no una cirugía y en qué momento realizarla, aún no está bien definida. Para decidir una conducta terapéutica en estos casos se deben considerar varios aspectos relevantes:
a. La posibilidad de desaparición de la malformación: No existe evidencia concreta y certera que demuestre la regresión completa de una malformación congénita pulmonar en período postnatal. Hay datos que han mostrado regresión de lesiones micro-quísticas y hay evidencia de que muchas malformaciones mejoran en el tiempo, pero la evidencia actual no permite asumir que ocurrirá una regresión completa ni permite establecer en qué casos se podría esperar esta evolución38,39,43
b. Evitar síntomas y complicaciones relacionados con la malformación: Está demostrado que los pacientes que han presentado síntomas o signos relacionados a la malformación pulmonar (hemorragia, infección, distress respiratorio, desplazamiento mediastino, etc.) previo a la cirugía poseen tasas de complicaciones tres veces mayores, tanto en complicaciones intraoperatorias como postoperatorias y requieren mayor tiempo de hospitalización en comparación a los pacientes que no los han presentado complicaciones. Esto se suma a que la presencia de complicaciones previo a la cirugía se asocia a una mayor tasa de conversión de cirugía mínimamente invasiva (toracoscopía) a cirugía abierta (toracotomía), aumentando así la morbilidad quirúrgica, las complicaciones en el manejo del dolor y las alteraciones relacionadas a una toracotomía. La complicación más frecuente de las malformaciones pulmonares congénitas y que empeora el pronóstico postoperatorio es la infección, cuya frecuencia va del 15 al 45% de los pacientes durante los dos primeros años de vida41,43,44.
c. Asociación con neoplasias: La asociación de malformación congénita pulmonar y la concurrencia con neoplasias como el Blastoma Pleuro-Pulmonar (BPP) y el Carcinoma Bronquio-Alveolar (CBA), entre otros, es conocida. Se ha demostrado que hasta un 5% de las Malformaciones Adenomatosas Quísticas pueden poseer elementos de BPP y hasta un 1% de CBA y el elemento más importante a considerar es que es imposible diferenciar a través de las imágenes (radiografía, TC o RM) cuales de estas malformaciones poseen o no elementos neoplásicos.37.
d. Crecimiento pulmonar: Diferentes estudios sugieren el beneficio de una resección temprana de una malformación pulmonar, dado el crecimiento pulmonar compensatorio que tendría el pulmón remante. Esto a diferencia de una cirugía más tardía que lo haría a expensas de una distensión alveolar.45
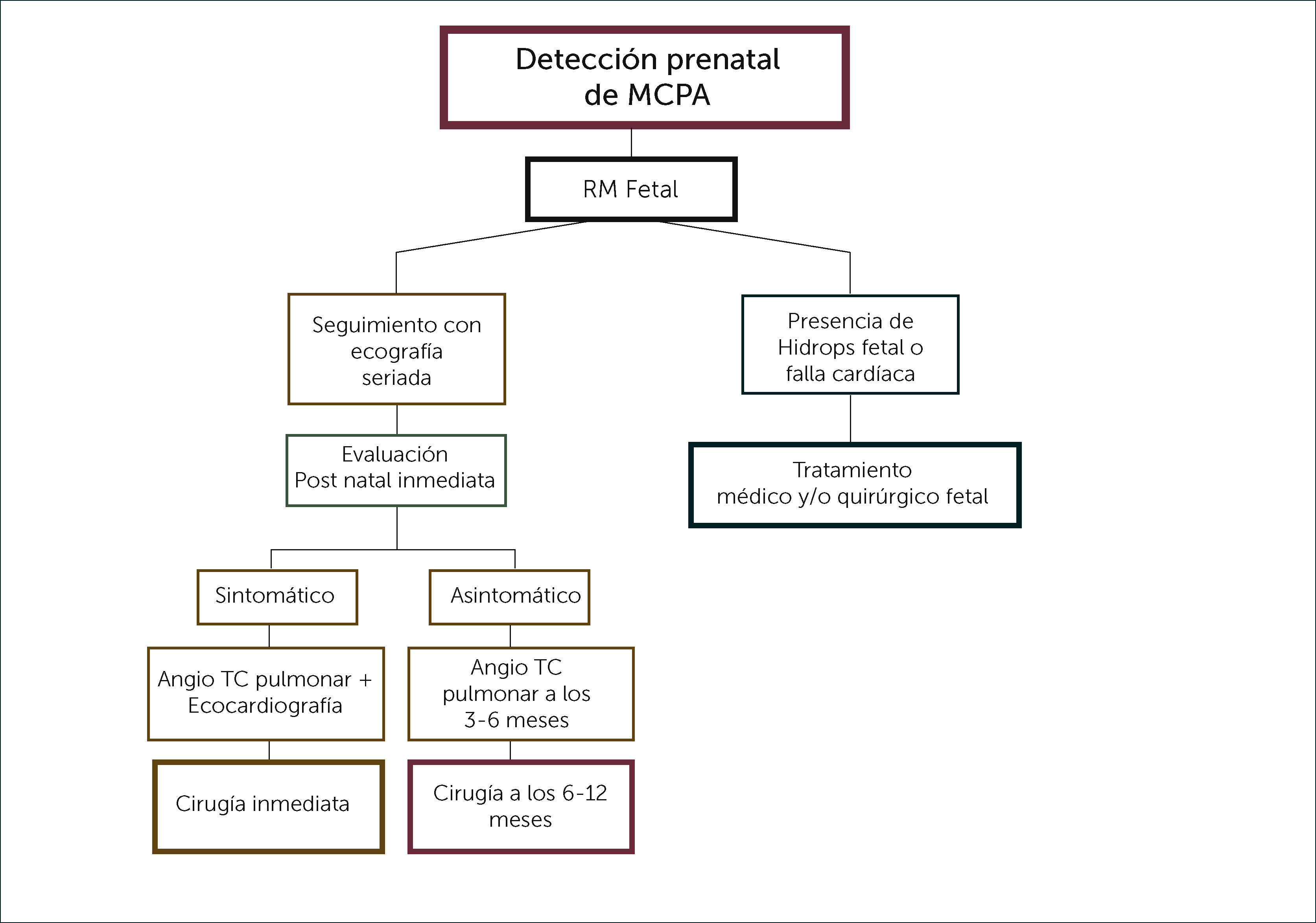
Actualmente no hay consenso acerca de cuánto tiempo debe observarse una MCPA en un paciente asintomático, cómo debe realizarse el seguimiento y en qué momento debe realizarse la resección quirúrgica. Considerando el aumento de la morbilidad por radiación, debido a que el seguimiento deberá realizarse con exámenes ionizantes (TC), el aumento de probabilidad de complicaciones con el paso del tiempo, la imposibilidad de identificar la presencia de neoplasias sólo con imágenes y el impacto en el crecimiento pulmonar de una resección temprana, hemos sugerido un protocolo de manejo de las MCPA en el periodo post-natal, que se detalla en la Figura 1.

Una vez tomada la decisión de operar a un paciente con MCPA, se debe decidir que tipo de abordaje quirúrgico y qué tipo de cirugía se va a realizar35,36,45.
Toracotomía o cirugía mínimamente invasiva (Toracoscopía)El abordaje al tórax hoy se puede realizar mediante una toracotomía o una toracoscopía. Los avances en instrumental quirúrgico hacen posible realizar una cirugía mínimamente invasiva en casi la totalidad de los pacientes que presentan una malformación pulmonar. Los beneficios de este tipo de abordaje son:
- -
Ausencia de morbilidad de una toracotomía
- -
Menor dolor post operatorio
- -
Ausencia de complicaciones de columna
- -
Mejor resultados estéticos (ausencia de cicatriz)
Dependiendo del tipo de malformación pulmonar, de su localización en 1 o más lóbulos y si es uni o bilateral, se decidirá por una lobectomía o una segmentectomía pulmonar.
CONCLUSIÓNLas malformaciones congénitas pulmonares y de vía aérea son patologías poco frecuentes cuyo diagnóstico prenatal ha aumentado. La mayoría de los casos evoluciona en forma satisfactoria, resultando en recién nacidos asintomáticos, sin embargo algunos de ellos presentan complicaciones graves y requieren de intervenciones precoces, incluso en período antenatal. Entender mejor la historia natural y la complejidad de estas malformaciones es de suma importancia para establecer definiciones y clasificaciones comunes y para poder realizar mejores protocolos de manejo, tanto prenatal como postnatal, para que estos pacientes lleguen a la resección de su malformación en las mejores condiciones y se logren los mejores resultados.
Los autores declaran no tener conflictos de interés, en relación a este artículo.