

Introducción
El empleo de diversos fármacos modificadores de la enfermedad (FAME) para el tratamiento de la Artritis Reumatoide (AR), ha demostrado su utilidad mejorando el curso y el pronóstico de ésta1. En la mayoría de los casos, estos medicamentos se han utilizado de forma intuitiva, sin conocer exactamente su mecanismo de acción2.
Durante los últimos 15 años se ha producido un gran avance en el conocimiento de la patogenia de la AR que, a su vez, ha derivado en un mejor conocimiento de los mecanismos de acción por los que los FAME son eficaces. Los datos de los que disponemos proceden tanto de estudios in vivo como in vitro. Sin embargo, su interpretación no es fácil. En el caso de los estudios in vivo es difícil establecer si el hallazgo descrito es una acción directa del fármaco o una consecuencia de otro mecanismo de acción diferente. Un ejemplo evidente es el de los nuevos tratamientos anti-TNF, cuyo mecanismo de acción es el bloqueo directo de esta molécula con la consiguiente disminución de la expresión de moléculas de adhesión y producción de moléculas inflamatorias como citocinas, quimiocinas, COX-2, etc. En cuanto a los estudios in vitro, o bien las dosis eficaces con frecuencia son mayores que las alcanzadas en plasma, o la relación tiempo-respuesta difiere de la observada en la práctica clínica. Esto nos impide determinar con exactitud si lo que estamos observando es un efecto terapéutico o tóxico como consecuencia de las altas dosis. Cuando ambos tipos de estudio llegan a la misma conclusión, por ejemplo que disminuya la expresión de moléculas de adhesión, es probable que estemos ante un verdadero mecanismo de acción del fármaco estudiado.
Teniendo en cuenta estas consideraciones, en esta revisión realizamos una puesta al día sobre los mecanismos de acción de los FAME basándonos en la información disponible sobre la patogenia de la artritis reumatoide.
Patogenia de la artritis reumatoide
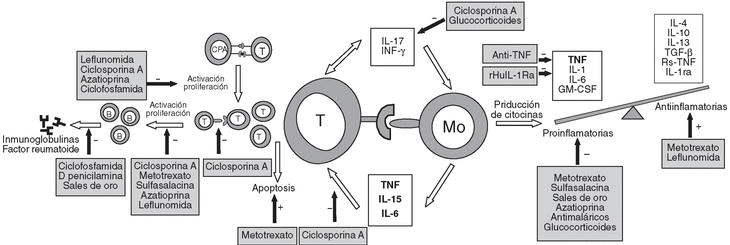
Es muy probable que, en un huésped genéticamente predispuesto, la interacción entre la célula presentadora de antígeno, que ha estado en contacto con un agente desencadenante desconocido, y su linfocito T específico sea determinante para el inicio de la enfermedad (fig. 1)3. Sin embargo, esta interacción podría preceder en mucho tiempo al inicio de los síntomas.

Figura 1. Patogenia de la artritis reumatoide I (ARI). Reconocimiento antigénico, proliferación celular y producción de citocinas. B: linfocito B; T: linfocito T; CPA: célula presentadora de antígeno; Mo: monocito; IL; interleucina; IFN-g: interferón g; TNF: factor de necrosis tumoral; rHuIL-1Ra: receptor de interleucina-1 humano recombinante; GM-GSF: factor estimulador de colonias de granulocitos; TGF-b: factor de crecimiento transformador b.
Conocemos mejor los fenómenos que perpetúan y amplifican la respuesta inflamatoria en la sinovial de pacientes con AR. La escasa producción de factores solubles por los linfocitos (principalmente INF-g, IL-18 e IL-17)4,5 y, sobre todo, la interacción celular entre linfocitos y macrófagos mediante las moléculas co-estimulantes expresadas6,7 conducen a la activación de los macrófagos y a la producción de una cascada de citocinas proinflamatorias (TNF, IL-1, IL-6, IL-15 y GM-CSF)8 entre las que, es bien conocido, el TNF juega un papel primordial (fig. 1).
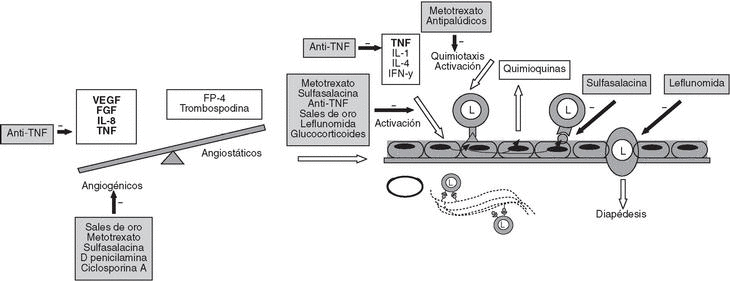
Esta producción de citocinas da lugar a la neovascularización9, la infiltración por leucocitos de la membrana sinovial y su migración a través de los vasos neoformados que expresan gran cantidad de moléculas de adhesión10 (fig. 2).

Figura 2. Patogenia de la artritis reumatoide II (ARII). Angiogénesis, activación del endotelio vascular y migración de leucocitos al foco inflamatorio. L: leucocito; VEGF: factor de crecimiento del endotelio vascular; FGF: factor de crecimiento fibroblástico; FP-4: factor plaquetario 4; IL. interleucina; TNF: factor de necrosis tumoral; IFN-g: interferón g.
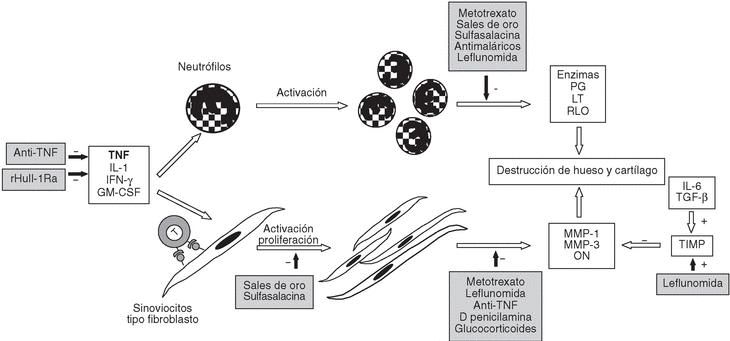
En este ambiente inflamatorio también se induce la proliferación de sinoviocitos que, junto con los neutrófilos llegados a la articulación, producen metaloproteasas, óxido nítrico, prostaglandinas, leucotrienos y radicales libres de oxígeno; todos ellos se implican en mayor o menor medida en la destrucción tisular que acompaña a la AR11 (fig. 3).

Figura 3. Patogenia de la artritis reumatoide III (ARIII). Mecanismos efectores. IL: interleucina; TNF: factor de necrosis tumoral; IFN-g: interferón g; GM-CSF: factor estimulador de colonias de granulocitos; PG: prostaglandinas; LT: leucotrienos; RLO: radicales libres de oxígeno; MMP: metaloproteasa de matriz; ON: óxido nítrico; TGF-b: factor de crecimiento transformador b; TIMP: inhibidor tisular de metaloproteasas.
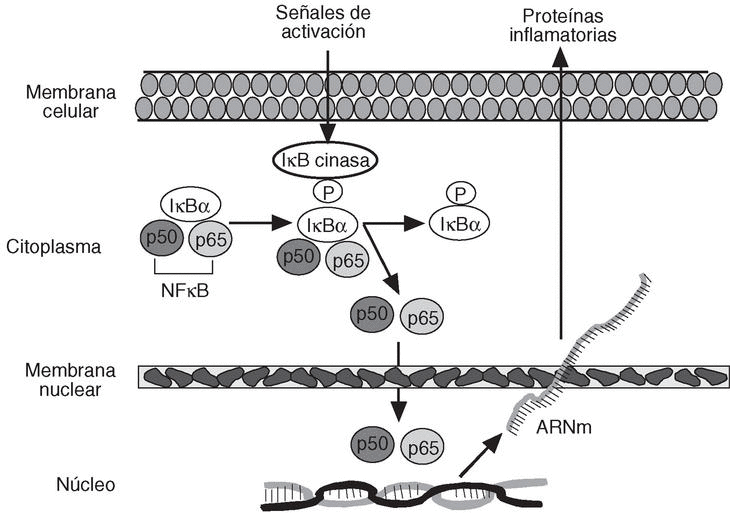
Un punto común de toda esta maraña de mecanismos patogénicos es que la producción de la mayoría de las moléculas relacionadas con la inflamación, como citocinas, quimiocinas, moléculas de adhesión, ciclooxigenasa-2 (COX-2), etc. esté controlada por factores de transcripción de la familia de NFkB (factor nuclear kB), aunque otros grupos de factores de transcripción como AP-1 o NFAT (factor nuclear de células T activadas) también están implicados12. NFkB está constituido por un dímero de proteínas situado en el citoplasma celular que, en condiciones basales, no activado, se encuentra ligado a su factor inhibidor IkB, lo que impide su entrada en el núcleo. Ante determinados estímulos, se produce la liberación de IkB por cinasas específicas; esto permite que NFkB entre en el núcleo celular activando las regiones promotoras de los genes de las distintas proteínas relacionadas con la inflamación (fig. 4). Así, por ejemplo, TNF e IFN-g, al actuar sobre sus receptores, activan la quinasa de IkB y permiten que NFkB induzca la transcripción y producción de todas las moléculas relacionadas con él.

Figura 4. Factores de transcripción. NFkB: factor nuclear kB; IkB: factor inhibidor kB; P: fosfato.
Metotrexato y Sulfasalacina
El metotrexato es un análogo de los folatos, capaz de inhibir la dihidrofolato reductasa, enzima implicada en la síntesis de novo de purinas y pirimidinas, necesarias para la formación de ADN y ARN que se requieren para la proliferación celular. Inicialmente, este fármaco se empleó pensando en su utilidad como antiproliferativo. Sin embargo, hay datos que no apoyan que éste sea su mecanismo de acción en la AR, como son: su eficacia a dosis más bajas que las empleadas en oncología, la rápida respuesta clínica y analítica tras su administración así como la ausencia de proliferación evidente de linfocitos en la sinovial reumatoide.
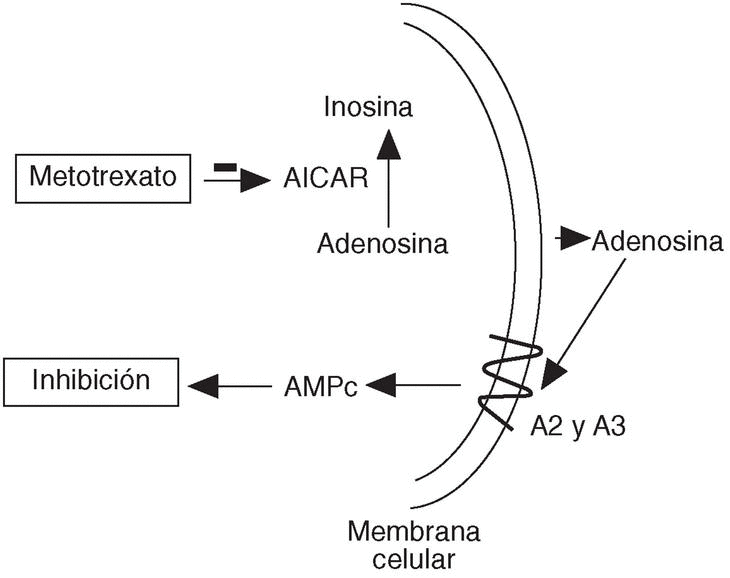
Actualmente se sabe que el metotrexato se acumula rápidamente en el interior de las células en forma de poliglutamatos, que tienen una alta afinidad por la 5-aminoimidazol-4-carboxiamida ribonucleótida (AICAR) transformilasa13. La inhibición de este sistema enzimático conduce a un aumento en la liberación de adenosina a la sangre14 (fig. 5). La adenosina interacciona con sus receptores específicos en células del sistema inmune, receptores tipo A2 y A315, desencadenando una respuesta inhibitoria, cuyas consecuencias son apoptosis de linfocitos activados16 y disminución de la producción de TNF, IFN-g, IL-6, IL-8 e IL-1217, así como aumento de la liberación de IL-6, IL-10 y del antagonista del receptor de IL-118 (fig. 1). Otras consecuencias de su acción son la inhibición de la migración de leucocitos (fig. 2), la reducción de la actividad COX-219 y la disminución en la producción de metaloproteasas20.

Figura 5. Mecanismo de acción del metotrexato. AICAR: 5-aminoimidazol-4-carboxiamina ribonucleótido transformilasa; AMPc: adenosín monofosfato cíclico.
La sulfasalacina comenzó a utilizarse para el tratamiento de la AR tras comprobar su eficacia en la artritis desarrollada por algunos pacientes con enfermedad inflamatoria intestinal. Sin embargo, parece que tiene un mecanismo de acción diferente en ambas enfermedades, ya que el 5-aminosalicilato, fragmento de la molécula de la sulfasalacina, es eficaz en la enfermedad inflamatoria intestinal pero no en la AR. En la actualidad sabemos que, al igual que el metotrexato, aumenta los niveles extracelulares de adenosina21 y produce así los mismos efectos.
Además, la sulfasalacina ejerce un potente efecto inmunomodulador sobre las células B inhibiendo su proliferación, lo que se traduce en hipogammaglobulinemia22 e inhibe la proliferación de células endoteliales23.
Leflunomida
A diferencia del resto de los FAME, el desarrollo de la leflunomida se ha dirigido desde un principio a su empleo en la AR. Es un profármaco que se transforma rápidamente en su metabolito activo, A77-1726, en el aparato gastrointestinal y el plasma. El A77-1726 inhibe la dihidro-orotato deshidrogenasa, limitando la síntesis de pirimidinas necesarias para la proliferación celular de los linfocitos T y B24. La disminución de los niveles de pirimidinas provoca la activación de la proteína p53, que se desplaza al núcleo celular impidiendo el paso de la fase G1 a la fase S del ciclo celular25. El hecho de que p53 se encuentre sólo en linfocitos activados explica el efecto selectivo de la leflunomida sobre estas células26 (fig. 1).
Sin embargo, en la sinovial reumatoide hay un número mínimo de linfocitos en proliferación, por eso diferentes autores han propuesto otras dianas terapéuticas para este fármaco. La leflunomida podría disminuir la actividad de las tirosincinasas27, suprimir la acción de NF*B28 y contribuir a la inhibición del contacto intercelular al disminuir la glicosilación de moléculas de membrana29. Todo ello conduciría a la disminución de la producción de citocinas proinflamatorias y aumento de las antiinflamatorias (fig. 1), inhibición del reclutamiento de leucocitos30 (fig. 2) y regulación del balance entre metaloproteasas y sus inhibidores (fig. 3).
Ciclosporina A y FK 506
La ciclosporina A es un potente inmunosupresor cuyo empleo ha supuesto un gran avance en el manejo de pacientes trasplantados. Debido a sus propiedades inmunomoduladoras, su uso se ha extendido a otras enfermedades en las que se produce una alteración de la inmunorregulación como es el caso de la AR, en la que ha demostrado su eficacia31.
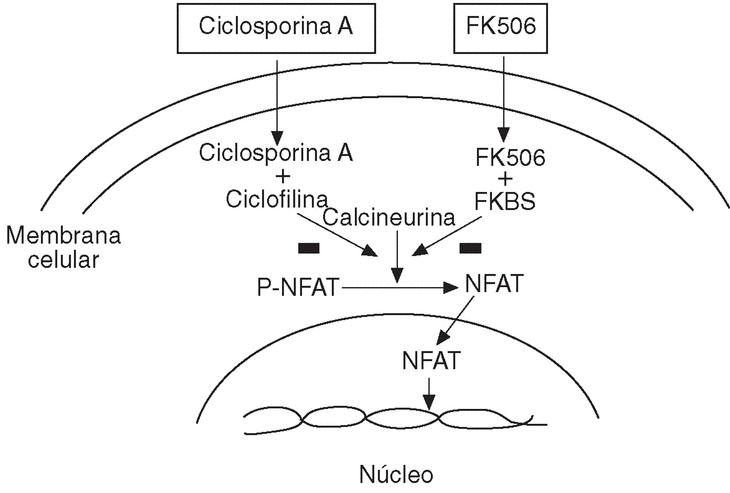
La ciclosporina A alcanza, por simple difusión, el citoplasma celular donde se une a las ciclofilinas. El complejo ciclofilina-ciclosporina A inhibe la calcineurina, enzima que defosforila el factor de transcripción NF-AT (fig. 6), presente en la mayoría de las células del sistema inmune. La defosforilación de este factor es esencial para su desplazamiento al núcleo celular donde activa la transcripción del gen de determinadas citoquinas (IL-2, IL-4, IL-8, GM-CSF, IFN-g, TNF) y de otras moléculas críticas para la respuesta inmune como CD40L32.

Figura 6. Mecanismo de acción de la ciclosporina A. NFAT: factor nuclear de células T activadas; P: fosfato.
Aunque la producción local de citocinas de origen linfocitario como IL-2, IL-4 e IFN-g es controvertida, Ziolowska et al han descrito que la ciclosporina A inhibe la producción de IL-17 mediada por IL-15 en linfocitos T del infiltrado sinovial33 (fig. 1). Además, otro mecanismo que explicaría la eficacia de este fármaco en la AR es su capacidad para inhibir la transcripción de genes de citocinas no linfocitarias o de moléculas linfocitarias coestimulantes. En este sentido, nuestro grupo ha estudiado su capacidad para inhibir la expresión de CD69 inducida por IL-15 en linfocitos34 (fig. 1).
El tacrolimus (FK506), fármaco con el que se han descrito casos aislados de eficacia en la AR, actúa de forma muy parecida a la CsA inhibiendo la actividad de la calcineurina, tras unirse a la proteína FKBPs (fig. 6) de la familia de las inmunofilinas35.
Otros fármacos modificadores de la evolución de la AR
Sales de oro
El efecto terapéutico fundamental de las sales de oro parece ejercerse sobre los neutrófilos36 y monocitos37, en los que se produce una acumulación selectiva del fármaco. La disminución de la producción de IL-1, IL-6, TNF e IFN-g es la consecuencia de su efecto sobre los monocitos2 (fig. 1). También se ha descrito que inhibe la proliferación de sinoviocitos y células endoteliales38 (fig. 2 y 3). En estas últimas se ha demostrado que inhiben la expresión de selectina E tanto in vivo como in vitro, lo que disminuye la migración leucocitaria hacia los focos de inflamación39,40 (fig. 2).
Por otra parte, parece que las sales de oro pueden inhibir directamente la unión del ADN a los factores de transcripción, como NFkB41, y disminuir la proliferación de sinoviocitos.
Antipalúdicos
El mecanismo de acción de los antipalúdicos sigue siendo desconocido. Se acumulan en las organelas celulares con un Ph ácido (lisosomas y aparato de Golgi) y, al afectar al Ph, pueden inhibir la acción de ciertas enzimas (proteasas, catepsinas y fosfolipasas). Esto provoca distintos efectos en los diferentes tipos celulares implicados en la inflamación: en los polinucleares inhibe la quimiotaxis y la fagocitosis42 (fig. 2); en los monocitos interfiere con el procesamiento y presentación del antígeno43 y en linfocitos inhibe la proliferación44.
D-penicilamina
Se han propuesto dos mecanismos de acción para la D-penicilamina. Por una parte, su molécula contiene un grupo tiol que podría alterar los receptores de la superficie celular mediante la reducción de grupos sulfidrilos45. Por otra, la oxidación de la D-penicilamina puede originar grupos peróxidos que afectarían a las distintas funciones celulares (linfocitos T, células endoteliales y fibroblastos)2.
Azatioprina
Es un análogo de purinas que interfiere con la síntesis de adenosina y guanina. Es un antimetabolito, tóxico para las células en fase S, que actúa inhibiendo la proliferación de las células T (46), B (47) y NK (48) (fig. 1). La azatioprina disminuye los niveles séricos de IL-6 aunque esto parece más consecuencia de la mejoría de la enfermedad que de una acción directa del fármaco49.
Ciclofosfamida y clorambucil
Ambos son potentes inmunosupresores que inhiben la proliferación celular en su fase premitótica (G2) además de distintas vías metabólicas. Alquilan el ADN, lo que se traduce en roturas, uniones cruzadas y disminución en su síntesis50. Su principal diana en la AR son los linfocitos B51 (fig. 1) aunque, a dosis altas, puede disminuir también la proliferación de linfocitos T, sobre todo CD8+52.
Glucocorticoides
Los glucocorticoides se unen, en el citoplasma celular, a receptores citoplasmáticos específicos que se expresan en distintos tipos celulares, incluyendo linfocitos, monocitos y neutrófilos. Son capaces de inhibir la actividad de NFkB aumentando la transcripción de su inhibidor IkB53. También regulan aspectos postranscripcionales como la traducción del ARN y la síntesis y secreción de proteínas.
Al aumentar los niveles de IkB, los corticoides impiden la internalización de NFkB en el núcleo celular inhibiendo la transcripción de los genes de COX-2, TNF, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IFN-g y GM-CSF54, moléculas de adhesión, etc. (fig. 1). La consecuencia de ello es un bloqueo rápido y generalizado de las funciones del sistema inmune.
Agentes biológicos
El mayor conocimiento de la patogenia de la AR junto con el avance experimentado por la biotecnología han permitido el uso de diferentes agentes biológicos dirigidos selectivamente contra algunos componentes de la respuesta inflamatoria sin causar inmunosupresión generalizada. Se han desarrollado agentes frente a moléculas de adhesión, quimiocinas, componentes del complemento, moléculas de activación y coestimulantes en la superficie de los linfocitos T, etc. Pero, sin duda, las que han experimentado un mayor avance han sido las dirigidas contra citocinas proinflamatorias, fundamentalmente IL-1 y TNF que, como hemos visto, modulan la mayor parte de los procesos que provocan inflamación y destrucción articular.
Antagonista del receptor de IL-1 (IL-1Ra)
El IL-1Ra es una citocina antiinflamatoria secretada por macrófagos que se une al receptor de la IL-1 pero sin transmitir ninguna señal activadora, de forma que, al interferir la unión de IL-1 con su receptor, impide su función55. Aunque se han detectado niveles valorables de IL-1Ra en el líquido sinovial de pacientes con AR, para producir la inhibición completa de IL-1 se requerirían niveles de IL-1Ra 10-100 veces superiores a los de ésta56. La producción de IL-1Ra humano recombinante (rHuIL-1Ra) ha permitido su uso clínico para bloquear los receptores de IL-1. La consecuencia de la administración de rHrIL-1Ra es la inhibición de la producción de prostaglandina E2 y colagenasa por los condrocitos evitando además la inhibición de la síntesis de glicoproteínas y la inducción de la degradación de proteoglicanos que produce la IL-157. Por estas razones, el tratamiento con rHuIL-1Ra en pacientes con AR ha demostrado que reduce el daño estructural58.
Anti-TNF
El TNF es quizá la citocina más relevante de las implicadas en la patogenia de la AR. El bloqueo de su actividad provoca la reducción de los niveles séricos de otros mediadores inflamatorios (IL-1 e IL-6)59,60, de la expresión de quimioquinas (IL-8 y MCP-1)61, de la migración linfocitaria62 y del factor de crecimiento del endotelio vascular (VEGF)63. Además, reduce los niveles séricos de metaloproteasas64 y óxido nítrico65 (figs. 1-3).
En la actualidad disponemos de 2 agentes biológicos anti-TNF: el infliximab, un anticuerpo monoclonal quimérico que se une al TNF soluble y al unido a la membrana celular; etanercept, una molécula diseñada por ingeniería genética que consta de 2 porciones extracelulares del receptor p75 del TNF unidas a la fracción Fc de IgG humana, para conferirle mejores propiedades farmacocinéticas. Los excelentes resultados clínicos obtenidos con estos fármacos han hecho que, hoy día, varios fármacos anti-TNF estén en distintas fases de desarrollo.
Conclusiones
En los últimos años, el conocimiento de la patogenia de la AR y los mecanismos de acción de los fármacos modificadores de su evolución han avanzado paralelamente. Estamos convencidos de que un conocimiento más profundo de cómo actúan los FAME en el complejo entramado de la AR nos permitirá hacer un uso más juicioso de estos fármacos y sus combinaciones.