



El síndrome de interrupción del tallo pituitario (PSIS) se caracteriza por la demostración neurorradiológica de un tallo pituitario ausente, interrumpido o hipoplásico, adenohipófisis aplásica/hipoplásica o neurohipófisis ectópica. Este síndrome se ha relacionado con formas severas de hipopituitarismo congénito (HPC), asociado a múltiples deficiencias de hormonas pituitarias (MPHD). Evaluamos a pacientes con HPC y PSIS, analizando los signos y los síntomas neonatales al diagnóstico, relacionándolos con las deficiencias hormonales pituitarias y signos neurorradiológicos presentes. Estudiamos retrospectivamente a 80 pacientes asistidos en el Hospital de Niños de Córdoba, con diagnóstico de HPC, de los cuales 42 (52%) presentaron PSIS; 22 mujeres y 20 varones, EC: 5 días-9,5 años. El 62% presentó MPHD y el 38% insuficiencia somatotrófica aislada (IGHD). El análisis de las variables perinatales demostró antecedentes de parto natural en el 52% (11/21) de las MPHD vs. 13% (2/15) de las IGHD. Cuatro pacientes, 2 con MPHD y 2 con IGHD presentaban antecedentes obstétricos consistentes en presentación podálica y transversa respectivamente, todos ellos resueltos mediante operación cesárea. Los signos y los síntomas perinatales fueron hipoglucemia: 61% en MPHD vs. 19% en IGHD, p: 0,0105; ictericia: 38% en MPHD vs. 25% en IGHD; micropene: 77% en MPHD y colestasis: 19% en MPHD. Convulsiones neonatales se presentaron en el 75% de los niños con MPHD e hipoglucemia. EC media de consulta: 2,1 años en MPHD (30% en el período neonatal, 70% antes de 2 años) y 3,6 años en IGHD (44% en menores de 2 años). Los pacientes con MPHD presentaban: tallo no visible 81% (n: 21/26) vs. tallo hipoplásico: 19% (n: 5/26), p: 0,0001; en IGHD 56% (n: 9/16) vs. 44% (n: 7/16), p: 0,5067, respectivamente. El 100% de los neonatos con HPC tenían tallo pituitario ausente. Concluimos que la demostración de PSIS en niños con HPC proporciona información valiosa como predictor de la severidad fenotípica, la presencia de MPHD y de la respuesta al tratamiento. La baja frecuencia de antecedentes obstétricos posicionales potencialmente distócicos, como parte de los mecanismos fisiopatogénicos responsables de PSIS, indicaría la necesidad de analizar la importancia de posibles factores genéticos y epigenéticos involucrados. El diagnóstico precoz de HPC debe sospecharse en presencia de signos y síntomas clínicos, tales como hipoglucemia, colestasis, micropene y defectos asociados en la línea media facial. La resonancia magnética cerebral debe formar parte de los estudios complementarios en pacientes con esta presunción diagnóstica, especialmente a edades tempranas. El reconocimiento tardío de esta entidad puede aumentar la morbilidad y la mortalidad con efectos potenciales deletéreos y permanentes.
Pituitary stalk interruption syndrome (PSIS) is characterised by the combination of an interrupted or thin pituitary stalk, absent or ectopic posterior pituitary, and anterior pituitary hypoplasia. It is manifested as isolated (IGHD) or combined pituitary hormone deficiencies (CPHD) of variable degrees and timing of onset, with a wide spectrum of clinical phenotypes. PSIS may be an isolated morphological abnormality or be part of a syndrome. A retrospective evaluation is presented of clinical signs and symptoms present at early life stages, as well as an analysis of their relationship with hormone laboratory tests and diagnostic imaging in children with congenital hypopituitarism (CHP), and PSIS. This study was performed in a single centre on a sample of 42 children out of a total of 80 CHP patients, with a chronological age range between 5 days and 9.5 years from a database analysed over a period of 26 years. The study included 26/42 (62%) with CPHD and 16/42 (38%) with IGHD. The analysis of perinatal variables showed a natural delivery in 52% (11/21) of CPHD vs 13% (2/15) of IGHD. Four patients, two with CPHD and two IGHD had breech and transverse presentation respectively. All of them were resolved by caesarean section. The perinatal histories showed hypoglycaemia (61% CPHD vs 19% IGHD, P=.0105), jaundice (38% CPHDvs25% IGHD), micropenis (75%CPHD), hypoglycaemic seizures (75% CPHD), and cholestasis (19% CPHD). The mean CA of consulting for CPHD patients was 2.1 years, 30% in neonatal period and 70% before 2 years. The mean chronical age (CA) was 3.6 years in IGHD patients, with 44% of them less than 2 years. MRI showed that 81% of CPHD patients had absence of pituitary stalk vs 19% with thin pituitary stalk (P=.0001); Patients with IGHD presented 56% absence of pituitary stalk vs 44% with thin pituitary stalk (P=.5067). All (100%) of the patients diagnosed in the neonatal stage had absent pituitary stalk. The characterisation of GH deficient patients by presence and type of hypothalamic-pituitary imaging abnormality provides valuable information as a predictor of phenotypic severity, treatment response, and the potential to develop additional hormonal deficiencies. We conclude that demonstrating PSIS in children with HPC provides valuable information as a predictor of phenotypic severity, presence of MPHD, and response to treatment. The low frequency of potentially dysfunctional positional obstetric history, as part of the pathophysiological mechanisms responsible for PSIS, would indicate the need to analyse the importance of possible genetic and epigenetic factors involved. Early diagnosis of HPC should be suspected in the presence of clinical signs and symptoms, such as hypoglycaemia, cholestasis, micropenis, and associated facial midline defects. MRI should be part of complementary studies in patients with this diagnostic suspicion, especially at an early age. Late recognition of this entity may increase morbidity and mortality with potential permanent deleterious effects.
El síndrome de interrupción del tallo pituitario (PSIS) se caracteriza por la demostración neurorradiológica de una tríada que incluye: tallo pituitario ausente, interrumpido o hipoplásico, adenohipófisis aplásica o marcadamente hipoplásica o neurohipófisis ectópica (EPP)1-4. Inicialmente fue descripta por Fujisawa et al. como transección del tallo pituitario5. La prevalencia reportada del PSIS no ha sido bien establecida; recientes publicaciones indican una incidencia estimada de 0,5/1.000.000 de recién nacidos6. El desarrollo anormal de la glándula pituitaria ha sido descripto con una frecuencia aproximada del 50% en pacientes con hipopituitarismo congénito (HPC)2. Este síndrome pertenece al espectro de las anomalías de la línea media cerebral y puede estar asociado a otras malformaciones extrapituitarias2,7,8.
Los mecanismos subyacentes implicados en la ontogenia del PSIS no han sido totalmente dilucidados. Lesiones producidas por injurias perinatales fueron sospechadas a partir de la demostración en estos pacientes de antecedentes de partos distócicos por presentación podálica y con hipoxia perinatal. Sin embargo, el hallazgo de un importante número de pacientes que no presentaban estos antecedentes, unido a la comprobación de casos familiares y a la presencia de otras malformaciones extrapituitarias asociadas, condujo a la formulación de nuevas hipótesis2,8-10. Un anormal desarrollo pituitario durante la embriogénesis ha sido propuesto, a partir de la observación de mutaciones en genes involucrados en el desarrollo neurológico temprano, tales como HESX1, LHX4, OTX2, SOX3 y PROKR211. Otros genes han sido identificados como responsables del PSIS, los cuales han sido vinculados con la holoprosencefalia, indicando que el PSIS forma parte del espectro fenotípico de esta entidad. Entre los genes involucrados podemos mencionar TGIF, GLI2, SHH, CDON, GPR 161 y PROKR212,13.
La baja frecuencia de mutaciones reportadas en niños con HPC, estimada en un 5% de los casos, permite hipotetizar que otras alteraciones y/o modificaciones en la expresión de genes relacionados contribuirían a esclarecer los mecanismos fisiopatogénicos determinantes de esta condición14.
En el período neonatal, el HPC puede manifestarse con signos y síntomas orientadores al diagnóstico, tales como hipoglucemia, convulsiones, ictericia prolongada, colestasis, micropene y excepcionalmente con la presencia de diabetes insípida1,2,15,16. En la infancia, la talla baja como consecuencia de la deficiencia de hormona de crecimiento (GH) (GHD) es la forma de presentación más común, seguida más tardíamente por la ausencia o retardo de desarrollo puberal. Entre un 40 y un 100% de los niños pueden presentar múltiples deficiencias de hormonas pituitarias (MPHD), responsables de otras sintomatologías asociadas3. Chen et al. revalorizaron la importancia de determinar las características anatómicas mediante resonancia magnética cerebral (RMC) de la región hipotálamo-pituitaria y su relación con la visibilidad del tallo pituitario y sus implicancias funcionales en niños con GHD y EPP. Si bien las bases fisiopatogénicas de la GHD en niños con PSIS y EPP continúan siendo investigadas, diferentes resultados indican que este síndrome se relaciona con formas más severas de presentación de la enfermedad asociada a MPHD3,17. Un diagnóstico tardío de la enfermedad determina una significativa morbimortalidad, lo que demuestra la importancia de identificar signos y síntomas clínicos presentes en etapas tempranas de la vida, especialmente en el período neonatal. La instauración de un tratamiento adecuado permitiría evitar el deterioro de la función cognitiva, los efectos deletéreos derivados de la MPHD y el abordaje apropiado de los defectos congénitos asociados2,16,18.
Una adecuada validación de la sintomatología neonatal y del comportamiento clínico derivado de la presencia de MPHD, relacionados con exhaustivas descripciones anatómicas, obtenidas por RMC, contribuiría a proporcionar una mejor caracterización de este grupo de pacientes y a esclarecer los posibles mecanismos fisiopatogénicos involucrados.
ObjetivosAnalizar a los pacientes con HPC y PSIS, evaluando los signos y los síntomas presentes en el período neonatal, en el momento del diagnóstico y relacionarlos con las deficiencias hormonales subyacentes y el comportamiento neurorradiológico observado.
Pacientes y métodosSe evaluó retrospectivamente a 80 pacientes con HPC diagnosticados en el Hospital de Niños de la Santísima Trinidad de Córdoba entre 1990 y 2016, de los cuales se analizaron 42 (52%) que presentaban PSIS, demostrado por RMC.
Criterios de inclusiónPacientes pediátricos con diagnóstico de HPC con la presencia de PSIS demostrado con RMC con gadolinio-ácido dietilentriamino-pentaacético (Gd-DTPA).
Criterios de exclusiónPacientes con diagnóstico posnatal de hipopituitarismo secundario a patologías tumorales, quirúrgicas o traumáticas demostradas.
Las variables clínicas consideradas se analizaron en el momento de la consulta y al inicio del tratamiento con GH: edad cronológica (EC) y datos antropométricos, relevándose de la historia clínica pertinente los antecedentes perinatales, los signos y los síntomas presentes en el período neonatal (tipo de parto, edad gestacional [EG], datos antropométricos, hipoxia, hipoglucemia, ictericia, colestasis, micropene, convulsiones, desórdenes visuales y malformaciones asociadas).
La talla y el peso fueron expresados en scores de desviaciones estándar (SDS) según tablas de población pediátrica argentina19, definiéndose como talla baja y/o peso un SDS menor o igual a –2 SDS20. Un score de Apgar menor de 7 fue considerado como distrés neonatal21.
Las variables bioquímicas utilizadas para el diagnóstico fueron: glucemia, bilirrubina, transaminasas, prolactina, cortisol, testosterona, hormona adrenocorticotropa, tirotrofina (TSH), tiroxina total (T4), tiroxina libre (T4 libre), hormona luteinizante (LH), folículo estimulante (FSH), hormona antidiurética, factor de crecimiento insulino símil tipo 1 (IGF1) y proteína transportadora 3 de IGF1 (IGFBP3).
Pruebas de estímulo: test de estimulación de TSH con factor hipotalámico liberador de TSH (TRH/TSH), test de estimulación de gonadotrofinas hipofisarias con factor hipotalámico liberador de gonadotrofinas (LH, FSH post-LH-RH) y test de estimulación de GH con arginina/clonidina.
En el período analizado, las determinaciones se realizaron empleando RIA e IRMA (inmunoensayos isotópicos DPC Diagnostic Products Corporation, Los Angeles, USA), IRMA (DSL Diagnostic Systems Laboratorios, Inc, Webster, Texas, USA), autoanalizador químico Modelo Hitachi 912 y EQLIA, electroquimioluminiscencia, Elecsys y Cobas (Roche Diagnostics GmbH, Mannhein, Alemania). La medición de IGF1 e IGFBP3 se realizó utilizando metodología IRMA (DSL Diagnostic Systems Laboratorios, Inc, Webster, Texas, USA; Access, Beckman Coulter, Prague, Czech Republic) y QLIA (quimioluminiscencia, Immulite 2000-Siemens Healthcare Diagnostic, United Kingdon).
Las líneas de corte consideradas para establecer el diagnóstico de deficiencia de las diferentes trofinas hipofisarias HPC se habían obtenido según las diferentes metodologías disponibles en el período contemplado en el presente estudio22-24. Se consideró GHD la demostración de GH basal en hipoglucemia < 10 ng/mL en neonatos25, niveles de IGF1 e IGFBP3 menores que –2 SDS para edad y sexo26 complementado con 2 pruebas de estímulo farmacológico para GH con arginina y clonidina con picos de GH máximos menores a 4,65 ng/mL para la metodología EQLIA, actualmente utilizada27. El diagnóstico de laboratorio de hipotiroidismo central se basó en valores bajos de T4 y bajos o en el rango normal de TSH según nuestros estándares de referencia. El test de TRH permitió distinguir un origen hipotalámico obtenidas respuestas de TSH sérica elevadas y sostenidas o hipofisario en ausencia de las mismas al factor liberador de TSH23.
Se consideró MPHD con la demostración de 2 o más ejes hormonales hipotálamo-pituitarios comprometidos.
Las RMC evaluadas habían sido efectuadas con resonadores de 0,5T inicialmente y de 1,5T en los últimos 7 años. Utilizamos un protocolo de RMC con especial valoración hipotálamo-pituitaria, con secuencias sagitales y coronales en T1-T2, centradas en la glándula pituitaria de 2mm de espesor, dinámicas con contraste paramagnético en planos coronal, y posteriormente sagital y coronal en T1 y 3D en T1 contrastados. Se realizaron además secuencias accesorias axiales en T2, FLAIR y difusión para evaluar el parénquima cerebral. Se consideró una adenohipófisis hipoplásica cuando era inferior a –2 DS, según referencias para edad y sexo28,29; tallo interrumpido cuando no era visible en su entera longitud y ausente cuando no era visible en su totalidad (pre y post-Gd-DTPA); tallo hipoplásico con dimensión menor a 3,25 ± 0,56mm a nivel del quiasma óptico y menor a 1,91 ± 0,4mm a nivel de la inserción en la glándula pituitaria30,31. La hiperseñal de la pituitaria posterior se determinó según su presencia y localización.
Análisis estadísticoSe realizaron pruebas de independencia de Fisher y odds ratio. Se consideró significación estadística p < 0,05. Se calcularon intervalos de confianza del 0,95 y pruebas t para comparar los grupos con GHD aislada (IGHD) y MPDH, para las variables de naturaleza continua: SDS de talla y SDS de talla media parental.
Los pacientes incluidos en el estudio prestaron su conformidad mediante consentimiento informado.
ResultadosDe los 42 pacientes con diagnóstico de PSIS, 22 fueron niñas y 20 varones (relación: 1,1), con edades comprendidas entre 5 días y 9,5 años. El análisis de las variables perinatales demostró antecedentes de parto natural en el 52% (11/21) de las MPHD vs. el 13% (2/15) de las IGHD. Cuatro pacientes, 2 con MPHD y 2 con IGHD, presentaban antecedentes obstétricos consistentes en presentación podálica y transversa respectivamente, todos ellos resueltos mediante operación cesárea. Los recién nacidos pequeños para la EG constituyeron el 21% (n: 9) del total de la muestra, 5 de ellos presentaron IGHD y 4 MPHD. El 14% (n: 6) del total de la población en estudio había cursado con un variable estado de distrés al nacimiento.
Los pacientes con HPC y MPHD representaron el 62% (n: 26), mientras que las formas de IGHD el 38% (n: 16).
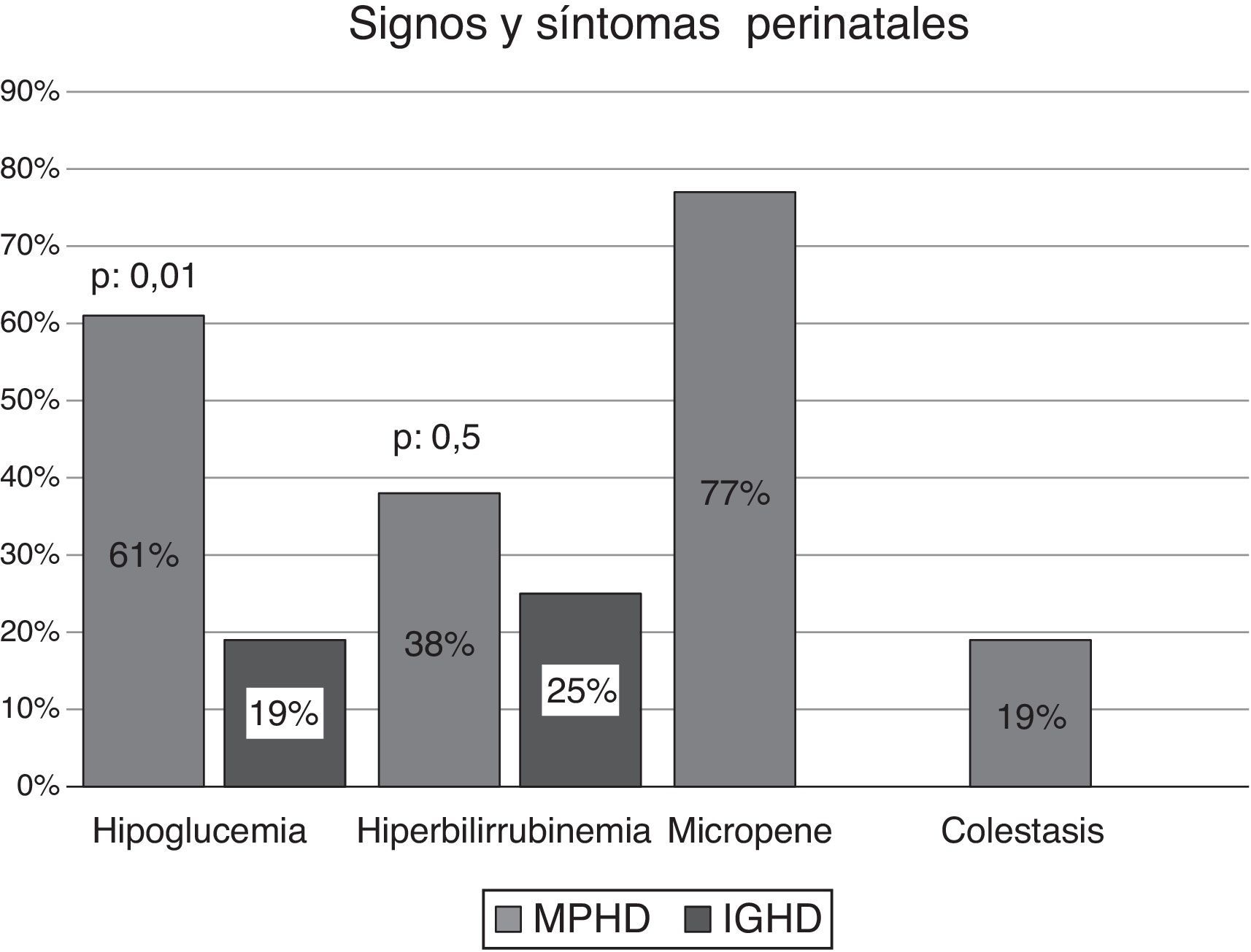
El análisis comparativo de las variables analizadas en las MPHD y en las IGHD demostró la presencia de hipoglucemia en el 61% (n: 16/26) de los pacientes vs. el 19% (n: 3/16) (p: 0,0105); ictericia en el 38% (n: 10/26) vs. el 25% (n: 4/16), respectivamente. Los pacientes con MPHD e hipoglucemia (n: 16) presentaron convulsiones neonatales en el 75% (n: 12/16). Solo en aquellos con MPHD se observó la presencia de micropene en el 77% (n: 9/12 varones); el 67% (n: 6/9) de ellos consultaron en el período neonatal y los restantes en el primer año de vida. El 19% (n: 5/26) registraba antecedentes de colestasis (fig. 1).

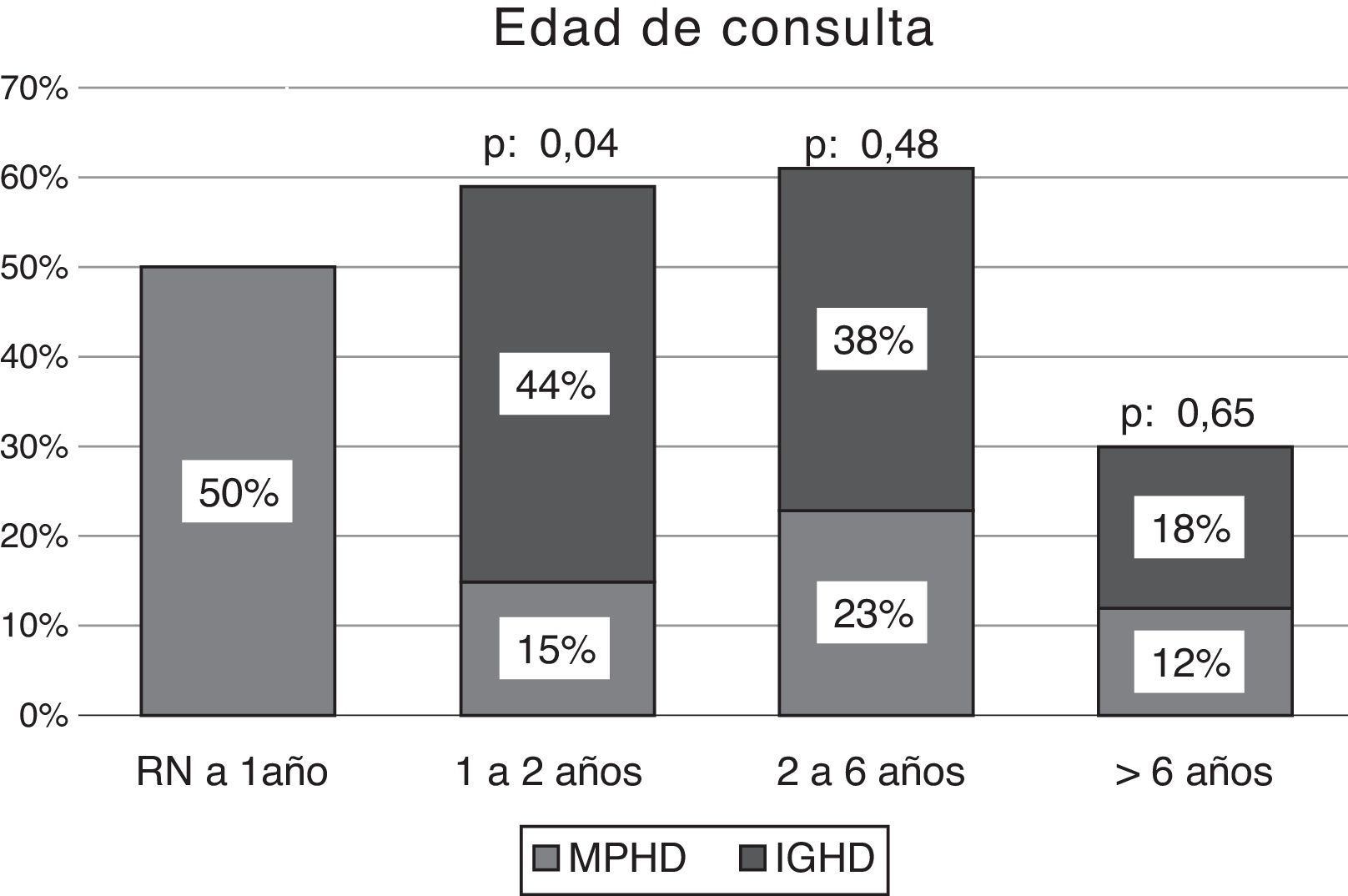
La EC media de consulta en los niños con MPHD fue de 2,1 años (rango: 5 días-9 años), el 31% (n: 8) de ellos consultó en el período neonatal y el 65% (n: 17) en los 2 primeros años de vida. En aquellos con IGHD, la EC media de consulta fue de 3,6 años (rango: 1-9,5 años), el 44% (n: 7) de ellos consultó antes de los 2 años de EC (fig. 2).

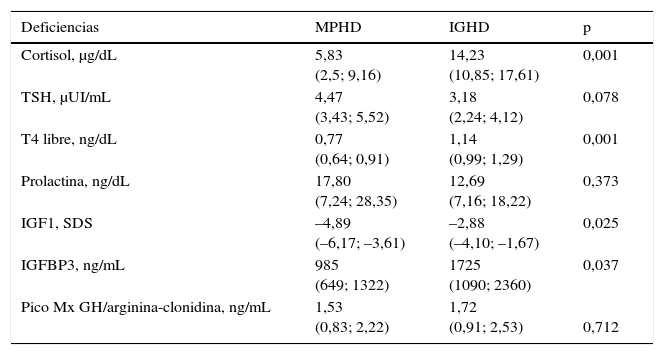
El 100% (n: 42) de los pacientes presentó GHD, el 24% (n: 10/42) de los niños fueron diagnosticados antes de los 2 primeros años de vida y solo 2 de ellos antes del primer año de vida. Los valores máximos de GH bajo estímulo farmacológico fueron de 1,57 ng/mL (0,83; 2,22) en MPHD y de 1,73 ng/mL (0,91; 2,53) en IGHD, p: 0,7123. Los niveles de IGF1 fueron significativamente menores en los niños con MPHD: –4,89 SDS (–6,17; –3,61) vs. aquellos con IGHD: –2,88 SDS (–4,10; –1,67); p: 0,0254. Un comportamiento similar se observó en los niveles de IGFBP3: en MPHD: 985 ng/mL (649; 1322) vs. en los IGHD: 1.725 ng/mL (1.090; 2.360); p: 0,0376 (tabla 1).
Niveles hormonales
| Deficiencias | MPHD | IGHD | p |
|---|---|---|---|
| Cortisol, μg/dL | 5,83 (2,5; 9,16) | 14,23 (10,85; 17,61) | 0,001 |
| TSH, μUI/mL | 4,47 (3,43; 5,52) | 3,18 (2,24; 4,12) | 0,078 |
| T4 libre, ng/dL | 0,77 (0,64; 0,91) | 1,14 (0,99; 1,29) | 0,001 |
| Prolactina, ng/dL | 17,80 (7,24; 28,35) | 12,69 (7,16; 18,22) | 0,373 |
| IGF1, SDS | –4,89 (–6,17; –3,61) | –2,88 (–4,10; –1,67) | 0,025 |
| IGFBP3, ng/mL | 985 (649; 1322) | 1725 (1090; 2360) | 0,037 |
| Pico Mx GH/arginina-clonidina, ng/mL | 1,53 (0,83; 2,22) | 1,72 (0,91; 2,53) | 0,712 |
Los valores se expresan como media (intervalo de confianza del 95%).
Los pacientes con MPHD demostraron un compromiso del eje tirotrófico en el 92% (n: 24/26) de los casos; adrenocorticotrófico en el 54% (n: 14/26); gonadotrófico en el 50% (n: 13/26) y deficiencia de hormona antidiurética en el 4% (n: 1/26). En relación con el compromiso del eje tirotrófico, la edad media al diagnóstico fue de 3 años (15 días-12,5 años), el 12,5% (n: 3/24) de ellos en el período neonatal. En aquellos con afectación del eje adenocorticotrófico, la edad media al diagnóstico fue de 3,2 años (rango: 5 días a 13 años), el 57% (n: 8/14) de ellos en el período neonatal. La única paciente con deficiencia de hormona antidiurética y severo compromiso adrenocorticotrófico, tirotrófico y de GHD, diagnosticada al mes de EC, falleció con posterioridad cercana al diagnóstico, en el marco de un espectro clínico de severidad que incluyó shock séptico y falla multiorgánica.
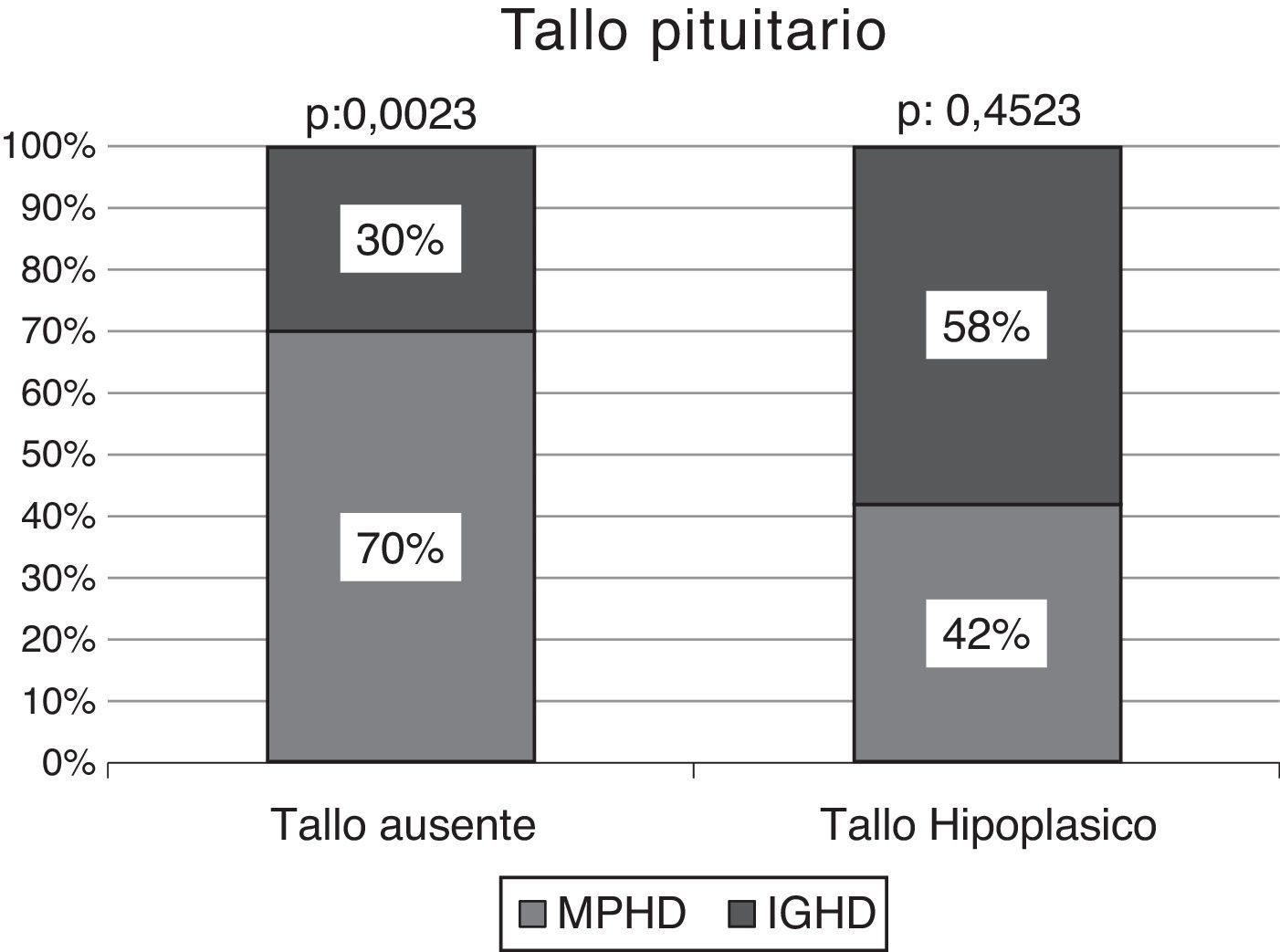
La RMC demostró un tallo no visible en el 71% del total de muestra (n: 30/42), resultando significativamente superior en pacientes con MPHD (70%, n: 21/30) vs. aquellos con IGHD (30%, n: 9/30); p: 0,0023 (fig. 3).

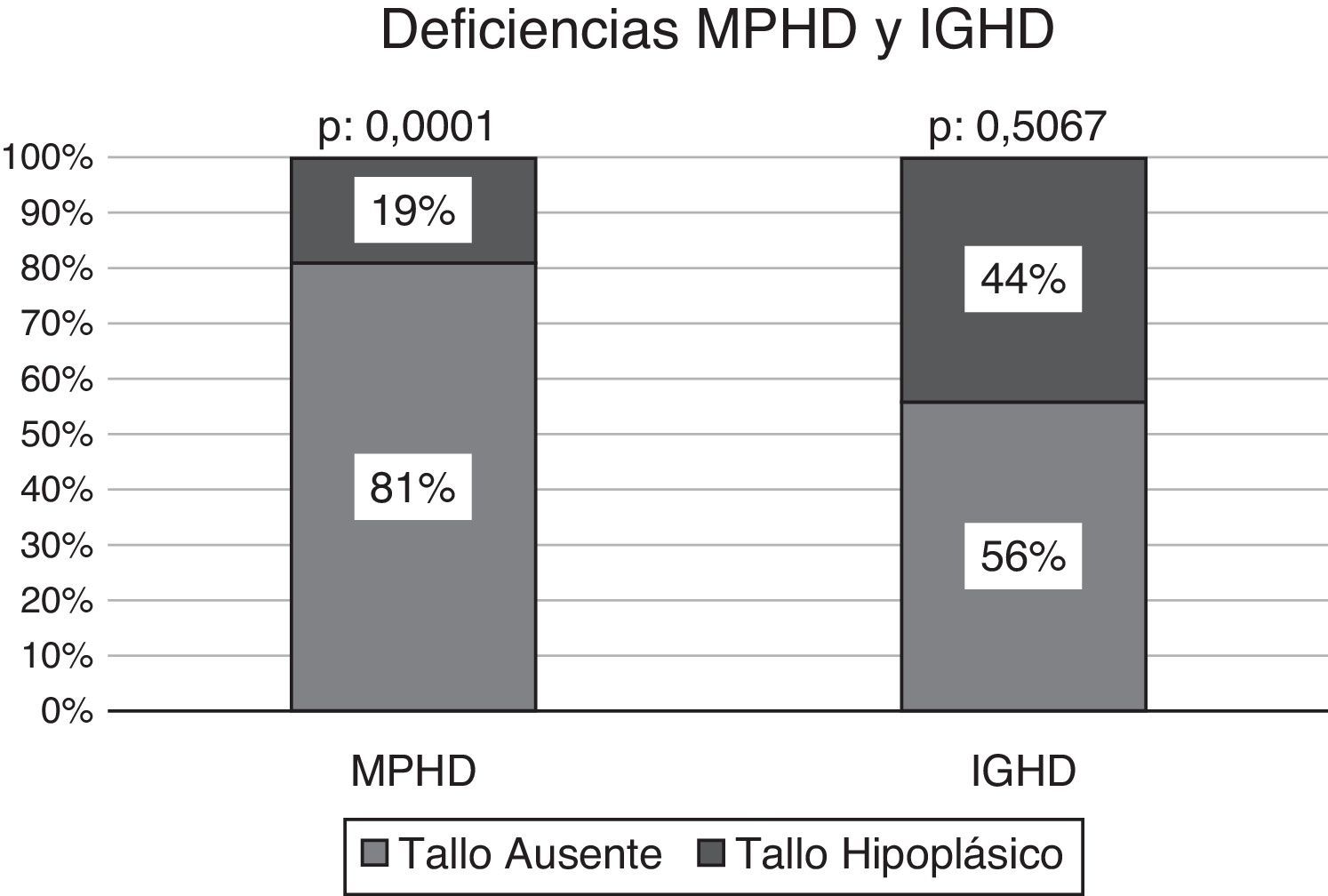
Efectuado el análisis comparativo de la presencia de un tallo no visible vs. tallo hipoplásico según el estado de deficiencia hormonal múltiple o aislada, los pacientes con MPHD presentaban un tallo no visible en el 81% de los casos (n: 21/26), significativamente superior a la condición de hipoplasia del mismo: 19% (n: 5/26); p: 0,0001. La frecuencia de un tallo no visible en los niños con IGHD se observó en el 56% (n: 9/16) vs. un tallo hipoplásico en el 44% (n: 7/16); p: 0,5067 (fig. 4).

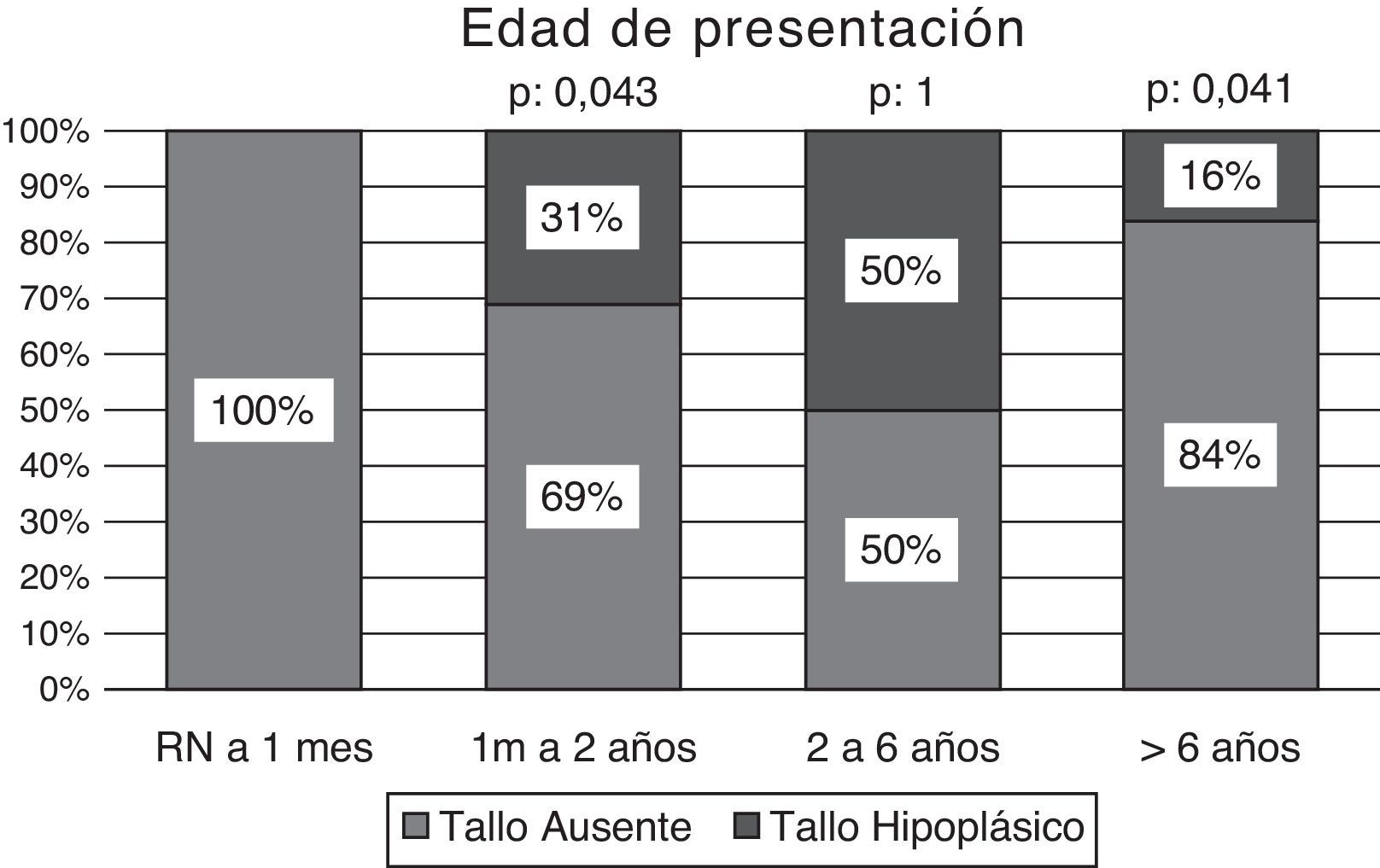
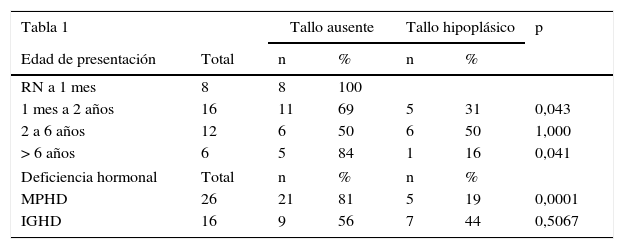
La totalidad de los pacientes (n: 8) diagnosticados en la etapa neonatal presentaron tallo pituitario ausente, resultando significativamente más frecuente en los 2 primeros años de vida comparado con la presencia de un tallo hipoplásico en este grupo etario (tabla 2) (fig. 5).
Edad de presentación y deficiencia hormonal según la visibilidad del tallo
| Tabla 1 | Tallo ausente | Tallo hipoplásico | p | |||
|---|---|---|---|---|---|---|
| Edad de presentación | Total | n | % | n | % | |
| RN a 1 mes | 8 | 8 | 100 | |||
| 1 mes a 2 años | 16 | 11 | 69 | 5 | 31 | 0,043 |
| 2 a 6 años | 12 | 6 | 50 | 6 | 50 | 1,000 |
| > 6 años | 6 | 5 | 84 | 1 | 16 | 0,041 |
| Deficiencia hormonal | Total | n | % | n | % | |
| MPHD | 26 | 21 | 81 | 5 | 19 | 0,0001 |
| IGHD | 16 | 9 | 56 | 7 | 44 | 0,5067 |

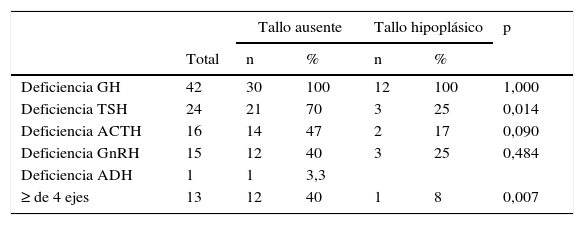
Analizado el comportamiento del tallo pituitario en relación con los ejes hormonales analizados, fue significativamente mayor la frecuencia de un tallo no visible en niños con compromiso del eje tirotrófico: 70% (n: 21/30) y en aquellos con afectación de 4 o más ejes hormonales: 40% (12/30) vs. la presencia de tallo hipoplásico 25% (n: 3/12; p: 0,0144) y 8% (n: 1/12; p: 0,007), respectivamente (tabla 3).
El PSIS se asoció a la presencia de pituitaria posterior ectópica en el 78% (n: 20/26) de los casos con MPHD vs. el 73% (n: 11/16) de aquellos con IGHD. Ambos grupos presentaron adenohipófisis hipoplásica en el 100% de la muestra (tabla 4).
Resonancia magnética cerebral y malformaciones asociadas
| Deficiencias | MPDH | IGHD | ||
|---|---|---|---|---|
| n | % | n | % | |
| Adenohipófisis hipoplásica | 26 | 100 | 16 | 100 |
| Neurohipófisis ectópica | 20 | 83 | 11 | 69 |
| Neurohipófisis ausente | 3 | 12,5 | 2 | 12,5 |
| Neurohipófisis normal | 1 | 4 | 3 | 18,7 |
| Malformaciones extrapituitarias asociadas | Hipoplasia del quiasma y nervios ópticos, ausencia septum pellucidum (n: 3) Malf. de Arnold-Chiari tipo 1 (n: 2). Hipoplasia del cuerpo calloso (n: 2) Labio leporino, fisura palatina (n: 1) Riñón en herradura (n: 1) | Atrofia cerebelosa (n: 1) Comunicación interauricular (n: 1) Estenosis pulmonar y hemivértebras T12 y L1 (n: 1) | ||
Analizada la presencia de malformaciones extrapituitarias asociadas, las mismas se observaron en el 28,5% (n: 12) de los niños con PSIS, las cuales estaban presentes en el 31% (n: 8/26) de los niños con MPHD vs. el 19% (n: 3/16) de IGHD. Evaluadas las malformaciones descriptas en niños con MPHD, 3 de ellos presentaban displasia septoóptica, caracterizada por ausencia de septum pellucidum, hipoplasia del quiasma y nervios ópticos, con el consiguiente compromiso de la agudeza visual; 2 agenesia de cuerpo calloso; 2 malformación de Arnold-Chiari tipo 1; un paciente labio leporino con fisura palatina y un niño riñón en herradura. En 3 pacientes con IGHD se demostró la presencia aislada de hemivértebras en T12 y L1, comunicación interauricular y atrofia cerebelosa (tabla 4).
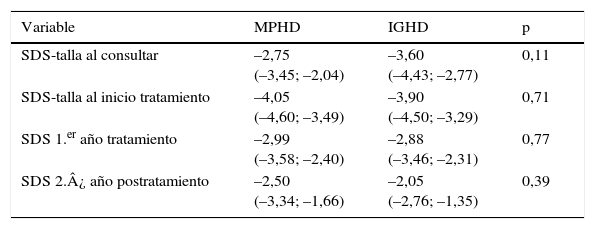
En los niños con MPHD, el tratamiento con GH se inició a la edad media de 4,5 años (6 meses-14,5 años) con una talla media inicial de –4,05 SDS (–1,8 a –6,3 SDS), observándose una ganancia de talla en el primer año de 1,06 SDS y de 0,5 SDS en el segundo año de tratamiento. Aquellos con IGHD iniciaron el tratamiento a los 5,4 años (17 meses-10,6 años) con una talla media de –3,9 SDS (–2,0 a –6,9 SDS), demostrándose una ganancia de talla en el primer año de 1,02 SDS y 0,83 SDS en el segundo año, sin diferencias significativas entre ambos grupos (tabla 5).
SDS de talla a la consulta, al inicio del tratamiento y evolución
| Variable | MPHD | IGHD | p |
|---|---|---|---|
| SDS-talla al consultar | –2,75 (–3,45; –2,04) | –3,60 (–4,43; –2,77) | 0,11 |
| SDS-talla al inicio tratamiento | –4,05 (–4,60; –3,49) | –3,90 (–4,50; –3,29) | 0,71 |
| SDS 1.er año tratamiento | –2,99 (–3,58; –2,40) | –2,88 (–3,46; –2,31) | 0,77 |
| SDS 2.¿ año postratamiento | –2,50 (–3,34; –1,66) | –2,05 (–2,76; –1,35) | 0,39 |
El 24% (n: 10/42) de los niños con GHD fueron diagnosticados antes de los 2 primeros años de vida y solo 2 de ellos antes del primer año de vida.
Discusión y conclusionesEn el presente estudio nosotros describimos las características clínicas, hormonales y neurorradiológicas en una cohorte de niños con PSIS, admitidos en un período de 26 años de práctica asistencial en un centro de endocrinología pediátrica.
La muestra obtenida se desprendió de una cohorte mayor de 80 niños con HPC, condición comúnmente subdiagnosticada, cuya incidencia es estimada en 4,2 por 100.000 por año, con una prevalencia de aproximadamente 45,5 por 100.00032,33.
En su etiopatogenia han sido señalados factores genéticos, medioambientales pre y posnatales o producto de una combinación de ambos34, de allí la importancia de relacionar, en una serie bien caracterizada de pacientes pediátricos, el fenotipo clínico, bioquímico, los defectos del neurodesarrollo subyacentes, y las manifestaciones propias de su asociación con una deficiencia aislada o múltiple de hormonas pituitarias.
El PSIS ha sido reportado en aproximadamente la mitad de los pacientes con HPC, aunque su exacta prevalencia es desconocida16. Nosotros examinamos 42 niños con esta condición, los cuales representan la mitad de los pacientes con HPC de nuestra serie, en concordancia con los datos reportados por Bar et al., obtenidos de un estudio longitudinal extendido por un período de 30 años2. A diferencia del mismo y de otros reportes, que reúnen un número similar de pacientes que señalan una relación masculino/femenina elevada (1,7 a 6,9)2-4,6, nuestros datos demuestran una frecuencia similar por sexo.
En relación con los posibles mecanismos responsables del PSIS, ha sido referida una incidencia variable de partos distócicos con presentación podálica, descripta en entre el 16 y el 90% de los casos1,2,4,6,15. Entre las hipótesis vertidas, y en concordancia con las anomalías estructurales pituitarias subyacentes, se ha indicado que las mismas serían producto de una alterada perfusión e isquemia pituitaria en grado variable, con o sin preservación del flujo sanguíneo vascular del tallo pituitario, que derivaría en mal rotación fetal e inadecuada presentación en el momento del parto4. En nuestra serie, solo 4 niños tenían antecedentes de mal rotación in utero, 2 de los cuales mostraban presentación podálica y 2 transversa, todos ellos nacidos por operación cesárea. Estas discrepancias señalarían que los mecanismos fisiopatogénicos subyacentes no están completamente dilucidados y la importancia de continuar analizando las implicancias de posibles factores genéticos y epigenéticos involucrados.
En el presente estudio nosotros describimos el espectro clínico-radiológico en relación con las deficiencias hormonales asociadas. Al respecto es importante señalar que los niños menores de un año presentaban formas severas de MPHD, destacándose entre los signos y los síntomas relevantes en el período neonatal la presencia hipoglucemia en el 61% de los neonatos, acompañada de convulsiones en el 75% de ellos, sumado a la demostración de micropene, hipocortisolismo e hipotiroidismo en el 67, el 57 y el 12,5% de los casos, respectivamente. Este fenotipo se asoció significativamente a ausencia de tallo pituitario, en concordancia con los datos proporcionados por la literatura2,5,7. Esto permitiría señalar el valor pronóstico de la integridad del tallo pituitario, obtenida post-Gd-DTPA, pudiéndose inferir que su interrupción se correlaciona con el número y magnitud de las deficiencias hormonales subyacentes que influyen en la severidad de los signos y los síntomas clínicos ya presentes en etapas tempranas de la vida1,3.
Nuestros datos indican que frecuentemente la sintomatología neonatal no es interpretada apropiadamente y las causas determinantes pueden ser esclarecidas tardíamente. Un cuidadoso relevamiento de síntomas y signos combinados, como hipoglucemia, ictericia, colestasis y micropene, durante el período neonatal y en la infancia temprana, puede orientar al estudio de un HPC. Si consideramos que el retraso de crecimiento puede no constituir un signo guion en esta etapa de la vida, la RMC con especial valoración de la región hipotálamo pituitaria debería formar parte de los protocolos de estudios complementarios frente al espectro clínico y de laboratorio descriptos. Pampanini et al. proponen la RMC como primera línea de investigación en la infancia temprana, basados en el análisis de 68 niños diagnosticados de GHD antes de la edad de 4 años, en los cuales se demostró la presencia de una anormalidad hipotálamo-pituitaria en el 83,8% de los casos y en el 100% de pacientes con MPHD7. Recientemente Kochi et al. reportan resultados falsos negativos en test de estímulos farmacológicos de GH, en pacientes con glándula pituitaria posterior ectópica, identificada con una RMC sagital simplificada y proponen esta práctica como método de investigación de primera línea en pacientes con HPC35.
En concordancia con otros reportes de la literatura2,6, nuestro trabajo demuestra la presencia de otras malformaciones extrapituitarias asociadas a PSIS, las cuales deberían ser cuidadosamente relevadas en el conjunto de estudios complementarios indicados a estos pacientes. Estos hallazgos indicarían además que las mismas podrían formar parte del espectro de anomalías hipotálamo-pituitarias desarrolladas durante la embriogénesis. La presencia de estas malformaciones y su posible relación con el espectro fenotípico de los niños afectados con PSIS contribuiría a orientar los estudios genéticos pertinentes.
En conclusión, nuestros resultados avalan que el diagnóstico precoz de HPC debe sospecharse en presencia de signos y síntomas clínicos, tales como hipoglucemia, colestasis, micropene y defectos asociados en la línea media facial. El reconocimiento tardío de esta entidad puede aumentar la morbilidad y la mortalidad con efectos potenciales deletéreos y permanentes.
Basados en el relevamiento de antecedentes perinatales en nuestra serie de pacientes, podríamos señalar que la baja frecuencia de antecedentes obstétricos posicionales potencialmente distócicos, señalados como parte de los mecanismos fisiopatogénicos responsables de PSIS, indicaría la necesidad de profundizar respecto a la influencia de posibles factores genéticos y epigenéticos involucrados. De allí la importancia de recomendar la realización de estudios de genética molecular acordes al fenotipo, que contribuyan a establecer el diagnóstico etiológico.
Nuestros resultados nos permitirían concluir que en niños durante la infancia temprana, con un espectro clínico y de laboratorio evocador de hipopituitarismo, la indicación de RMC debería formar parte de los protocolos de estudios complementarios pertinentes.
Nuestras observaciones nos permiten inferir que la demostración de PSIS en pacientes con hipopituitarismo proporciona una información valiosa para el médico y la familia del paciente como predictor de la severidad fenotípica, la respuesta terapéutica esperada y el potencial compromiso de otras trofinas pituitarias, las cuales pueden manifestarse en el curso evolutivo de esta condición.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no poseer conflictos de interés.
Agradecimientos a las licenciadas Laura González, Sandra Cejas, Celina Tettamanti, por su colaboración, y a la Fundación Endocrinológica Infantil Córdoba, por su respaldo para la realización del presente trabajo.