





INTRODUCCIÓN
El síndrome de Turner es la cromosomopatía asociada al sexo más común entre las mujeres1. Se caracteriza por la ausencia total o parcial de un cromosoma X en todas o en parte de sus células. En el tipo más común, presente hasta en el 60% de las pacientes, existe una monosomía completa del cromosoma X en línea única. Con elevada frecuencia, el cariotipo es un mosaicismo, fundamentalmente 45X/46XX2,3. La tasa de mosaicismos depende de la cantidad de tejidos estudiados; cuando se analiza uno solo se observan mosaicos en el 25-30% de las pacientes, pero cuando se estudian 2 esta tasa asciende al 65%3. Las modernas técnicas citogenéticas, como la fluorescence in-situ hybridization (FISH) o la reacción en cadena de la polimerasa (PCR) permiten también detectar más eficazmente los mosaicos frente a las técnicas convencionales4. En el resto de los pacientes el síndrome es debido a anomalías estructurales del cromosoma X en línea única (isocromosoma de los brazos largos, deleciones, cromosomas en anillos) o aberraciones del cromosoma Y.
Es una entidad clínica bien definida posnatalmente caracterizada por estatura baja, fenotipo peculiar (cuello alado, cubitus valgus, tórax ancho con mamilas muy separadas, facies triangular, metacarpianos cortos), disgenesia gonadal y disfunción ovárica. Son frecuentes las anomalías cardiovasculares, presentes en el 15-30% de las pacientes estudiadas postnatalmente5-7. Los defectos más comunes son la válvula aórtica bicúspide (14-19%) y la coartación de aorta (4-10%). El pronóstico es bueno en la mayoría de los casos detectados posnatalmente, y el desarrollo intelectual suele ser normal. Las carencias hormonales se pueden solventar con terapia hormonal sustitutiva, que se inicia antes de la edad adulta1.
En vida fetal, la expresividad de este síndrome es muy diferente. Los hallazgos ecográficos más típicos son la translucencia nucal muy aumentada y el higroma quístico, acompañados con frecuencia de edema subcutáneo, derrame en alguna cavidad serosa fetal o incluso de una hidropesía fetal, detectables todos ellos en la primera mitad de la gestación8-10. Esta presentación fetal del síndrome se acompaña de un pronóstico mucho peor que la forma posnatal, y su mortalidad intrauterina desde el primer trimestre hasta el término de la gestación se encuentra próxima al 80-90%11. Esto explica que la incidencia de la monosomía X al nacimiento sea mucho menor (1/1.500-2.500 niñas nacidas vivas) que su incidencia en vida fetal (se estima que afecta al 3% de los embriones femeninos concebidos)1,12.
El objetivo de este estudio es comparar las características prenatales de esta entidad, principalmente el tipo de cromosomopatía y la incidencia y tipo de defectos cardíacos con las observadas posnatalmente y analizar la evolución habida tras el diagnóstico prenatal.
PACIENTES Y MÉTODOS
Para la realización de este trabajo hemos recuperado de nuestro archivo las 51 gestaciones en que se realizó diagnóstico prenatal de síndrome de Turner en el período entre 1990 y 2003. En 45 casos la monosomía X era completa en línea única (88%), en 5 casos (10%) eran mosaicismos 45X/46XX y 1 tenía una doble aneuploidía 46X,+21. La edad materna media fue 30 años (desviación estándar [DE] 5,6; rango, 14-42 años). En 12 casos la edad era >= 35 años (23%). La edad gestacional media al diagnóstico fue 14 semanas (DE 2,6; rango 10-20 semanas).
La obtención de la muestra fetal para el estudio citogenético se llevó a cabo en 32 casos mediante amniocentesis (63%), en 11 casos con biopsia corial (22%) y en 8 casos se utilizó la sangre fetal obtenida mediante funiculocentesis (15%). Todos los casos diagnosticados mediante biopsia corial eran monosomías X en línea única. En 46 de los 51 casos el motivo del estudio citogenético fueron las anomalías detectadas en la ecografía del primer trimestre (en 10, además, la edad materna era >= 35 años y en 2 existían antecedentes de cromosomopatía). En 2 casos el estudio citogenético se llevó a cabo al observar anomalías en la ecografía del segundo trimestre. En otros 2 casos, la indicación fue la edad materna >= 35 años y en el restante el antecedente de enfermedad de Duchenne: en ninguno de éstos se observaron anomalías en el momento de la obtención de la muestra. En el caso de los mosaicismos, en 4 casos la indicación del estudio citogenético fueron los hallazgos ecográficos (en 2, además, la edad materna era >= 35 años) y sólo en este estudio se hizo por edad materna >= 35 años, sin anomalías ecográficas.
En la ecografía del primer trimestre, el hallazgo extracardíaco más común fue el higroma quístico cervical caracterizado por la existencia de una saculación líquida septada, bilobulada, localizada en la parte posteroinferior de la nuca fetal: estaba presente en 36 fetos y en 28 de ellos se acompañaba de hidropesía fetal, definida por la combinación de edema subcutáneo > 5 mm y derrame en al menos una cavidad serosa10. En otros 9 fetos existía una translucencia nucal aumentada, definida en nuestro medio como la colección econegativa subcutánea en la nuca fetal >= 3 mm13, entre las semanas 11 y 14 de gestación. En estos fetos con monosomía X el grosor medio de la translucencia fue de 6,5 mm (DE 2,6; rango, 3-10 mm). Se acompañaba de hidropesía en 3 casos. En un caso el hallazgo extracardíaco precoz fue un onfalocele. En 2 fetos las anomalías extracardíacas se detectaron en el segundo trimestre (engrosamiento nucal > 6 mm y leve acortamiento de huesos largos en ambos).
En 3 casos no se detectaron anomalías ecográficas antes del diagnóstico de la monosomía, pero tras éste sí se observaron en 2 de ellos: en un feto se apreció un edema plantar bilateral y en otro una malformación de Dandy-Walker y un defecto perimembranoso del septo interventricular de 3 mm. Sólo en un caso no se apreciaron anomalías morfológicas antes ni después de conocer el cariotipo, que correspondió a un feto 45X/46XX.
En 37 casos los padres optaron por la interrupción voluntaria del embarazo (IVE) (73%) y en 13 se produjo la muerte intrauterina espontánea del feto (25%); ésta tuvo lugar siempre en la primera mitad de la gestación, y todos los fetos, excepto uno, tenían colecciones líquidas retronucales en sus diferentes formas. Sólo en 1 caso la gestación alcanzó el término, y nació una niña con un edema plantar bilateral, ya detectado en la ecografía prenatal, y con una exploración cardíaca normal.
En nuestra unidad la exploración ecocardiográfica fetal se reserva para las gestantes con alto riesgo de cardiopatía congénita fetal. Este riesgo elevado se establece por la presencia de factores maternos, como exposición a teratógenos, diabetes pregestacional o hijos previos con cardiopatía, y/o propiamente fetales, como la existencia de anomalías morfológicas extracardíacas, diagnóstico de cromosomopatía o alteración de la estructura cardíaca en la exploración básica. La ecocardiografía precoz (12-16 semanas) se realiza de forma combinada, transvaginal y transabdominal, y después de la semana 16 es siempre transabdominal. Se realiza una completa exploración segmentaria incluyendo la imagen de «4 cámaras», tractos de salida, arcos aórtico y ductal y retorno venoso sistémico utilizando imagen bidimensional complementada con Doppler pulsado convencional y color. En los últimos 29 fetos de la serie se exploró también la onda de flujo en el ductus venoso que, en condiciones normales, debe mantener un perfil anterógrado durante todo el ciclo cardíaco.
De los 51 fetos de la serie, en 12 casos no pudo realizarse ecocardiografía, al ser abortos ya en la primera ecografía (9 casos) o por ser diagnósticos muy tempranos al principio de la década de los noventa, cuando aún no se realizaba ecocardiografía precoz. Por tanto, el número de casos válidos para ecocardiografía fue de 39. En 36 de ellos las anomalías extracardíacas motivaron la exploración cardíaca, que precedió al diagnóstico citogenético. En los 3 restantes el estudio cardíaco se hizo tras conocer el diagnóstico citogenético: en ninguno de éstos se observaron signos ecográficos indicativos de cromosomopatía en el primer trimestre.
Las ecografías fueron realizadas con los ecógrafos Acuson XP128-10 (MontView, California), General Electric Logic 400 y Logic 500 (GE Medical Systems, Milwaukee, Wisconsin) con sondas entre 3 y 7 MHz. Para el estudio estadístico utilizamos un paquete básico (Stat-view 512, Microsoft).
Las variables continuas se describen como media y DE. Las variables categóricas se describen como distribución de frecuencias absolutas y categóricas. La significación estadística de la comparación de proporciones se obtiene con la prueba de la *2 o la prueba exacta de Fisher.
RESULTADOS
La edad gestacional media en la que se hizo la ecocardiografía fue 14 semanas (DE 2,7; rango, 12-20 semanas). En la mayoría de los casos (79%, 31/39) se hizo en el primer trimestre, entre las semanas 11 y 16 (media = 13 semanas, DE 1,7). La exploración fue satisfactoria ya en la primera visita en todos ellos, salvo en un feto examinado en la semana 11 y al que hubo que repetir la ecocardiografía 1 semana más tarde. En los 8 casos restantes la ecocardiografía se hizo en el segundo trimestre, a una edad gestacional media de 19 semanas (DE 0,7; rango, 18-20 semanas). En 5 de ellos su indicación fue la existencia de anomalías extracardíacas en la ecografía del segundo trimestre: en 2 casos no se había realizado ecografía en el primer trimestre y en los otros 3 ésta era normal, pero se había realizado muy precozmente (9-10 semanas). En los 3 restantes la ecocardiografía se hizo tras conocer el diagnóstico de monosomía X: en todas las ecografías previas se informaron como normales.
De los 39 fetos, 34 tenían la monosomía X en línea única (87%), 4 eran mosaicismos 45X/46XX, y el restante tenía una doble aneuploidía 46X,+21. Se detectó la existencia de una cardiopatía congénita en 15/39 fetos (39%), diagnóstico que precedió al citogenético en 14/15 (93%). La edad gestacional media al diagnóstico fue 14 semanas (DE 2,7; rango, 12-20 semanas) con 13 casos diagnosticados ¾ 16 semanas (87%). El diagnóstico más común fue la asimetría entre ambos ventrículos y las grandes arterias, con dominancia de las estructuras derechas e hipoplasia difusa del arco aórtico, sugestiva de la existencia de una coartación de aorta: estaba presente en 8/39 fetos (21%) (figs. 1a y b). En segundo lugar, el defecto más común fue el síndrome de ventrículo izquierdo hipoplásico, detectado en 5 fetos (13%) (fig. 2). En un caso se diagnosticó un defecto perimembranoso del septo interventricular de 3 mm, y en otro feto, un defecto completo del septo atrioventricular. Este último corresponde al feto con la doble aneuploidía 46X,+21. En los 4 fetos con mosaicismos la exploración cardíaca fue normal. En la tabla 1 se resumen los hallazgos ecocardiográficos de nuestra serie y su relación con el cariotipo fetal.

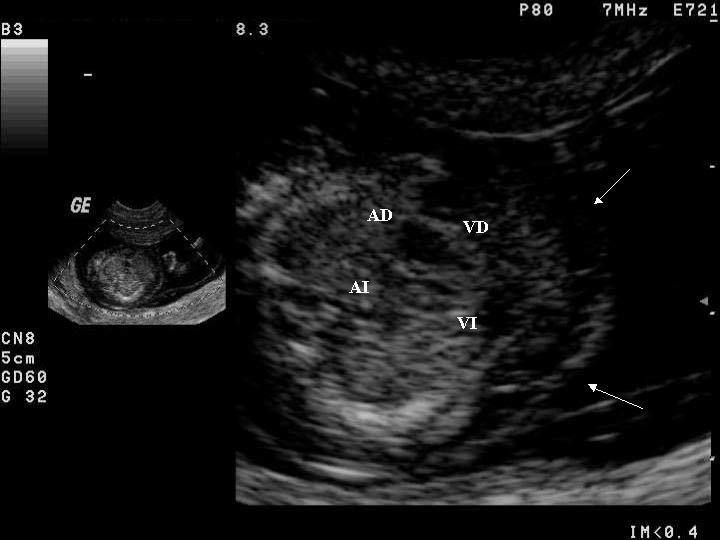
Figura 1. a) Imagen de "cuatro cámaras" de un corazón normal en la semana 14 de gestación, y b) imagen de "cuatro cámaras" de un corazón con asimetría de cavidades sugestiva de coartación, en la semana 13 de gestación. VI: ventrículo izquierdo; VD: ventrículo derecho; AI: aurícula izquierda; AD: aurícula derecha. Las flechas señalan el edema subcutáneo.


Figura 2. Imagen de "cuatro cámaras" de un corazón con hipoplasia de cavidades izquierdas, en la semana 13 de gestación. En la exploración con Doppler color se confirmó la ausencia de flujo transmitral y transaórtico. VI: ventrículo izquierdo; VD: ventrículo derecho; AI: aurícula izquierda; AD: aurícula derecha. Las flechas señalan el edema subcutáneo.

En 14 casos fue posible repetir la ecocardiografía al menos una vez más (rango, 1-4), semanas más tarde (media = 2; rango, 1-5). En 8 casos la nueva exploración confirmó lo apreciado en la anterior y en 4 se diagnosticó la muerte fetal. En los 2 casos restantes, con una primera ecocardiografía en la semana 11 (en uno satisfactoria e informada como normal y en el otro insatisfactoria) se observaron 1 semana más tarde signos ecográficos de coartación de aorta. En 1 de estos fetos se realizaron 2 exploraciones más con intervalos de 2 semanas, con los mismos hallazgos cardíacos; el feto falleció finalmente en la semana 18.
No observamos diferencias significativas en la tasa de cardiopatías en función de la presencia o no de hidropesía fetal; en 10 de los 27 fetos hidrópicos se detectó una cardiopatía (37%) y en 5 de los 12 no hidrópicos (42%) (p = 1,0). Por el contrario, cuando estudiamos la relación de las cardiopatías con la onda de flujo en el ductus venoso observamos que, cuando ésta era normal, la tasa de cardiopatías era menor (2/11, 18%) que cuando el flujo diastólico era ausente o reverso coincidiendo con la contracción atrial (11/18, 61%) (p = 0,05).
Del grupo de 39 fetos con monosomía X a los que se pudo realizar ecocardiografía, en 33 casos los padres optaron por la interrupción voluntaria del embarazo (IVE) (85%). En 5 casos se produjo la muerte intrauterina espontánea del feto (13%), entre las semanas 14 y 20 de gestación. En el caso restante, con monosomía X en línea única diagnosticada en la semana 14 y en el que no se apreciaron anomalías en la ecografía, los padres optaron por continuar con la gestación. En los controles sucesivos únicamente se observó un edema plantar bilateral, y la ecocardiografía se mantuvo normal. La exploración clínica del recién nacido confirmó los hallazgos prenatales; la exploración cardíaca posnatal también fue normal. De los 4 fetos con mosaicismo, en 3 los padres optaron por la IVE y el otro falleció intraútero de forma espontánea en la semana 20 de gestación.
Disponemos de comprobación neonatal o anatomopatológica en 26 de los 39 fetos (67%). En los restantes 13 casos no se dispone de estudio necrópsico por la multifragmentación fetal poslegrado (9 casos) o porque la IVE se llevó a cabo en centros sin recursos suficientes para un examen fetal precoz (4 casos). En 9 de los 15 fetos (60%) en que se detectó una anomalía cardíaca fue posible el estudio anatomopatológico, que confirmó los hallazgos prenatales, mientras que en los 6 restantes no disponemos de estudio post mortem. De los 24 fetos con ecocardiografía normal disponemos de seguimiento neonatal o anatomopatológico en 17 casos (71%): en todos ellos, el examen cardíaco fue normal.
DISCUSIÓN
La evolución de las pacientes con síndrome de Turner diagnosticadas en la vida posnatal, aceptablemente buena en la mayoría de los casos, contrasta notablemente con el pronóstico de los casos detectados prenatalmente1,14-16. En éstos es frecuente la muerte espontánea intrauterina, fundamentalmente cuando cursan acompañados de colecciones líquidas nucales y más aún si existe hidropesía fetal17,18. Esto queda probado también en nuestra serie. Estas pérdidas fetales espontáneas pudieran ser incluso mayores si no se detuviera su presumible evolución natural con la IVE. A la evolución gestacional tan adversa observada en nuestra serie, y en otras15,16, contribuye decisivamente el hecho de que la mayoría de los diagnósticos citogenéticos prenatales de monosomía X se realizan tras la detección de alguna anomalía en la ecografía. A diferencia de lo expresado por otros autores1, que consideran que la mayoría de los síndromes de Turner se diagnostican casualmente al realizar amniocentesis por la edad materna avanzada, en nuestra serie la mayoría de los casos se diagnosticaron en madres jóvenes y tan sólo en 3 casos (6%) no existía indicación ecográfica para realizar el estudio citogenético. El pronóstico de estos fetos puede ser muy diferente del apreciado en nuestra serie, procedente de un hospital terciario con un elevado índice de patología fetal.
Frente a lo publicado en series pediátricas, prenatalmente hemos observado diferencias cuantitativas y cualitativas en la asociación de cardiopatías al síndrome de Turner. En nuestra serie la tasa de cardiopatías es superior a la apreciada en los casos detectados posnatalmente (el 39 frente al 22-26%)5-7. Estas diferencias pueden ser incluso mayores si tenemos en cuenta que sólo la tercera parte de las pacientes con síndrome de Turner están incluidas en las series pediátricas. La mayoría de los casos no están diagnosticados, y corresponden a genotipos con mosaico o anomalías estructurales del cromosoma X5. La expresividad clínica de éstos es mucho menor, incluso nula, y la tasa de cardiopatías es muy inferior, lo que reduce globalmente la prevalencia de anomalías cardiovasculares en las pacientes con este síndrome.
En cuanto a la distribución de las cardiopatías, se diagnosticó una coartación de aorta en el 21% de los casos y un síndrome del ventrículo izquierdo hipoplásico en el 13%, frente al 4-10% y el 1-2%, respectivamente, observados posnatalmente5,6,19. Estas diferencias también han sido observadas por otros autores15,20,21. Ambas entidades son cardiopatías que no sólo tienen un momento de aparición variable sino que además se caracterizan por tener un marcado carácter evolutivo in utero22,23. De hecho, en un feto de nuestra serie se diagnosticó una coartación de aorta 1 semana después de haber informado otra exploración como normal. Por ello, es posible pensar que la tasa observada de cardiopatías en nuestra serie pudiera ser incluso mayor si no se detuviera su evolución natural por la IVE, en muchos casos realizada muy precozmente, con lo que se acentuarían las diferencias con los estudios posnatales. Además, aunque la tasa de seguimiento post mortem puede considerarse elevada en comparación con la de otras series15 y con una coincidencia diagnóstica plena tanto en los fetos con cardiopatía como sin ella, no disponemos de estudios necrópsicos en el 33% de los casos; éste es otro argumento más para pensar que quizá la tasa observada de cardiopatías pudiera ser incluso mayor.
En las series posnatales, clásicamente se ha considerado que la coartación de aorta era la cardiopatía más común en las pacientes con síndrome de Turner24. Sin embargo, este concepto tiene el sesgo derivado de la dificultad para la detección de la enfermedad leve de la válvula aórtica, con frecuencia asintomática, y más aún en el comienzo de la era ecocardiográfica. Series recientes y muy amplias han demostrado que la válvula aórtica bicúspide es la enfermedad cardíaca más común (9-34%) en las pacientes diagnosticadas posnatalmente5-7,25,26. Éste es un defecto que no suele diagnosticarse prenatalmente y que no fue detectado en ningún feto de nuestra serie. Del mismo modo, el drenaje venoso pulmonar anómalo parcial, que es más común en estas pacientes que en el resto de la población7, tampoco fue observado en ningún feto de nuestra serie.
Existen también diferencias en las cromosomopatías observadas en estas pacientes al comparar su distribución pre y posnatal. En nuestra serie, la mayoría de los fetos (87%) tenía la monosomía 45X en línea única, y el resto eran mosaicismos. Nuestra distribución es similar a la observada en otras series15,16,27. Por el contrario, en las series posnatales la distribución es diferente: Mazzanti et al6 tienen un 54% de cariotipos 45X, un 13% de mosaicismos y un 33% de anomalías estructurales, y para Gotzsche et al5 estas tasas son del 58, el 35 y el 7%, respectivamente. Es decir, en la vida fetal son mayoritarios los síndromes de Turner con cariotipo 45X en línea única, mientras que la contribución de los mosaicismos y las anomalías estructurales del cromosoma X es mayor posnatalmente. Estas diferencias permiten explicar la diferente evolución de los casos pre y posnatales. Los síndromes debidos a mosaicismos o anomalías estructurales serían en muchos casos silentes desde el punto de vista ecográfico fetal y, por tanto, quedarían sin detectar. De ahí su mayor impacto en las series posnatales. Por el contrario, la expresividad ecográfica de los cariotipos 45X en línea única es mucho mayor. Esto facilitaría su detección prenatal y explicaría su elevada letalidad intrauterina. Por ello, la prevalencia posnatal de este cariotipo es menor, a lo que contribuye, además, el efecto de las IVE. En una serie de 100 fetos con síndrome de Turner, se observó que la probabilidad de que el feto alcanzara el término de la gestación era mayor cuando su cariotipo era mosaico que cuando era 45X en línea única11. La elevada tasa de IVE en nuestra serie y el reducido número de fetos con mosaicismo impide extraer conclusiones en este apartado, aunque puede destacarse que ninguno de estos tenía cardiopatía.
Los comentarios anteriores enlazan directamente con la estrecha relación existente entre el tipo de cromosomopatía y la gravedad de la cardiopatía. Está probado en series posnatales que en las pacientes con síndrome de Turner en línea única, al igual que son más frecuentes las dismorfias típicas del síndrome, es más probable la existencia de un defecto cardíaco y que éste, además, sea más grave que en las pacientes con mosaicismo o anomalías estructurales del cromosoma X5,6. En una serie amplia de 179 pacientes con síndrome de Turner procedente de un estudio multicéntrico danés, se diagnosticó una cardiopatía congénita en el 38% de las pacientes con monosomía X en contraste con el 11% de las pacientes con mosaico. En ninguna de las pacientes con anomalías estructurales del cromosoma X se observaron anomalías cardiovasculares5. En la serie italiana, también multicéntrica, existía coartación de aorta en el 10,9% de las pacientes con monosomía X en línea única frente al 5,4% de los casos con mosaico y el 1% de las pacientes con anomalías estructurales en el cromosoma X6. En estos últimos, la anomalía más común es la válvula aórtica bicúspide7. Estos datos se confirman en nuestra serie: se diagnosticó una cardiopatía en el 41% de los fetos con monosomía X en línea única y no se observó ninguna en los fetos con mosaicismo.
La relación existente entre la onda de flujo en el ductus venoso y las cardiopatías en el primer trimestre de la gestación se ha demostrado28. Nuestra serie confirma que la tasa de cardiopatías es superior cuando en la ecografía del primer trimestre se observa una onda anómala del flujo en el ductus venoso y fundamentalmente la presencia de flujo ausente o reverso durante la contracción atrial (el 61 frente al 18%; p = 0,05). Por ello, el estudio de la onda de flujo en este vaso es obligado en todo feto con anomalías ecográficas en el primer trimestre. No observamos ninguna relación entre las cardiopatías y la hidropesía, probablemente debido a la etiología multifactorial de ésta en los fetos con síndrome de Turner10. Por otra parte, nuestra serie pone de manifiesto de nuevo la elevada capacidad diagnóstica de la ecocardiografía que permitió detectar todas las cardiopatías de nuestra serie, con una elevada tasa de seguimiento necrópsico. De la misma manera, confirmamos la eficacia de la ecocardiografía precoz para el diagnóstico en el primer trimestre de las cardiopatías graves. Esta técnica diagnóstica, que es altamente dependiente del explorador, ha de ser reservada para las gestaciones con alto riesgo de cardiopatía fetal. Entre éstas están aquellas en las que existen anomalías morfológicas fetales extracardíacas y, fundamentalmente, colecciones líquidas retronucales29,30. Por ello, siempre que se detecten anomalías fetales en el primer trimestre de la gestación debe recomendarse la realización de una ecocardiografía precoz que permitirá detectar, en manos expertas, un alto número de cardiopatías fetales, tal y como sucede en nuestra serie. En la monosomía X, dada su elevada letalidad intrauterina, la ecocardiografía precoz tendría además un especial valor al diagnosticar las cardiopatías graves antes de que se produjera el óbito fetal. Esto explica que en nuestra serie la tasa observada de cardiopatías en estos fetos sea superior a la apreciada en otras series fetales en las que no se aplicó ecocardiografía precoz16.
En conclusión, la mayoría de los síndromes de Turner diagnosticados prenatalmente tienen la monosomía X en línea única y casi todos tienen anomalías ecográficas que conducen al diagnóstico. En comparación con las series posnatales, la tasa de cardiopatías en estos fetos es mayor y los defectos son más graves. Esto justifica el mal pronóstico que acompaña al diagnóstico prenatal de esta cromosomopatía. La ecocardiografía fetal es altamente eficaz para el diagnóstico de las cardiopatías. Los fetos con esta cromosomopatía y con alteración en la onda de flujo del ductus venoso tienen mayor probabilidad de presentar cardiopatía.