




La enfermedad inflamatoria intestinal (EII) es una entidad común que puede afectar hasta un 0,37% de la población. La enfermedad de Crohn (EC) y la colitis ulcerosa (CU) son los dos tipos más importantes y se asocian a dolor abdominal, rectorragias, diarrea, signos de malnutrición y pérdida de peso1. Tanto la EC como la CU pueden presentar afectación extraintestinal en un 20-40% de los casos (tabla 1)2–5, estando implicados en su patogenia mecanismos genéticos e inmunes2,4. Se relacionan con la actividad de la EII: eritema nudoso, epiescleritis y artritis periférica4. La afectación cutánea en EII es frecuente (10% aproximadamente)6,7. Se han descrito numerosos hallazgos dermatológicos asociados, entre los que destacan, por su frecuencia, el eritema nudoso y el pioderma gangrenoso (tabla 2).
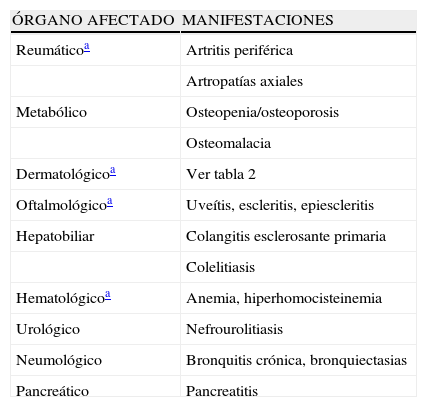
Manifestaciones extraintestinales de la enfermedad inflamatoria intestinal
| ÓRGANO AFECTADO | MANIFESTACIONES |
| Reumáticoa | Artritis periférica |
| Artropatías axiales | |
| Metabólico | Osteopenia/osteoporosis |
| Osteomalacia | |
| Dermatológicoa | Ver tabla 2 |
| Oftalmológicoa | Uveítis, escleritis, epiescleritis |
| Hepatobiliar | Colangitis esclerosante primaria |
| Colelitiasis | |
| Hematológicoa | Anemia, hiperhomocisteinemia |
| Urológico | Nefrourolitiasis |
| Neumológico | Bronquitis crónica, bronquiectasias |
| Pancreático | Pancreatitis |
Modificado de: S. Larsen, K. Bendtzen, O. H. Nielsen, Ann Med 2010; 42: 97–114.
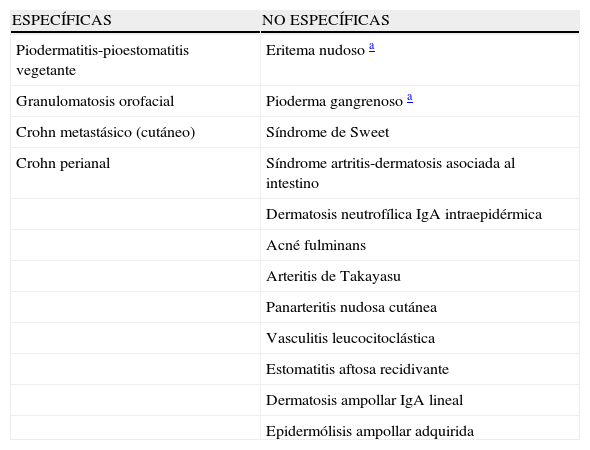
Dermatosis relacionadas con la enfermedad inflamatoria intestinal (EII). La aparición de estos procesos puede preceder a la EII
| ESPECÍFICAS | NO ESPECÍFICAS |
| Piodermatitis-pioestomatitis vegetante | Eritema nudoso a |
| Granulomatosis orofacial | Pioderma gangrenoso a |
| Crohn metastásico (cutáneo) | Síndrome de Sweet |
| Crohn perianal | Síndrome artritis-dermatosis asociada al intestino |
| Dermatosis neutrofílica IgA intraepidérmica | |
| Acné fulminans | |
| Arteritis de Takayasu | |
| Panarteritis nudosa cutánea | |
| Vasculitis leucocitoclástica | |
| Estomatitis aftosa recidivante | |
| Dermatosis ampollar IgA lineal | |
| Epidermólisis ampollar adquirida |
Existen procesos cutáneos específicos de la EII que son importante reconocer, ya que pueden preceder a la EII y ser la única manifestación identificable del proceso4,8. Por tanto, todos los pacientes diagnosticados de EII deben ser explorados dermatológicamente. En su mayoría no se correlacionan de forma consistente con la actividad de la EII (excepto el eritema nudoso4,6) ni predicen su pronóstico9. En este trabajo se revisa la etiopatogenia, la clínica, la histopatología y el tratamiento de cada entidad en relación con la EII. Asimismo, se ha prestado especial atención a la actualización terapéutica con fármacos inmunosupresores y biológicos en pacientes que asocian el proceso cutáneo y el digestivo.
Dermatosis específicasPiodermatitis-pioestomatitis vegetanteLa piodermatitis-pioestomatitis vegetante (PPV) es una rara inflamación mucocutánea crónica10. Aunque puede aparecer antes11, está siempre asociada a EII y se considera un marcador específico del proceso digestivo7,12. Es más frecuente en CU que en EC12,13. Predomina en hombres y puede aparecer a cualquier edad (es más frecuente entre los 20 y los 59 años)10. La piodermatitis vegetante afecta la piel y la pioestomatitis vegetante asienta sobre la mucosa oral (lo más frecuente), vaginal, nasal y ocular10. Aunque en la literatura en ocasiones aparecen descritos por separado, se considera que se trata de diferentes espectros de la misma entidad12. Su patogenia se desconoce. Sin embargo, se han implicado factores inmunológicos y microbiológicos13.
En las mucosas las lesiones suelen ser múltiples y están formadas por papulopústulas estériles de coloración blanca o amarillenta sobre una base eritematosa difusa. En ocasiones adoptan un patrón lineal en «huella de caracol»7. Pueden confluir formando grandes placas vegetantes y friables que desembocan en erosiones dolorosas y úlceras13. La mucosa bucal (pioestomatitis vegetante) tiene un aspecto eritematoso, engrosado y granular, debido a la presencia de numerosas erosiones superficiales13. Se ha descrito la ulceración de la epiglotis y la laringe. La piodermatitis vegetante puede aparecer antes o después que la afectación oral10 y también se caracteriza por papulopústulas sobre base eritematosa que confluyen para formar placas vegetantes. La distribución es asimétrica y predomina en cuero cabelludo y pliegues (axilares e inguinales)10. También es posible su localización en cara, tronco y parte distal de extremidades. El cuadro puede ir acompañado de fiebre y adenopatías submandibulares. Existe eosinofilia en sangre periférica hasta en el 90% de los casos10.
En el estudio histológico pueden apreciarse abscesos intraepiteliales y/o subepiteliales formados por abundantes neutrófilos y eosinófilos10. Se acompaña de hiperplasia pseudoepiteliomatosa, acantosis, hiperqueratosis y áreas de disociación intraepitelial. En la lámina propia papilar existe un infiltrado inflamatorio agudo y crónico compuesto por eosinófilos, linfocitos, neutrófilos y células plasmáticas. En ocasiones puede haber un discreto infiltrado inflamatorio perivascular superficial. El estudio de inmunofluorescencia (IF) es negativo10. El diagnóstico se establece mediante la correlación entre los hallazgos clínicos, la negatividad de los cultivos de las pústulas, la relación con EII, la eosinofilia periférica y el estudio histológico. El diagnóstico diferencial debe realizarse especialmente con el pénfigo vegetante de tipo Neumann o de tipo Hallopeau10, por lo que puede ser de ayuda el estudio de IF.
Como tratamiento mediante la enfermedad oral, se usan corticoides sistémicos acompañados de terapia tópica basada en antisépticos y corticoides de potencia alta. Puede combinarse con azatioprina, sulfametoxipiridazina y ciclosporina con éxito. La dapsona es una posibilidad alternativa. Recientemente, el infliximab, junto a terapia de mantenimiento con metrotexato en EC asociada a pioestomatitis vegetante, ha obtenido buenos resultados14. El tratamiento específico de la EII es fundamental7,10.
Dermatosis específicas de CrohnGranulomatosis orofacialEs la afectación del labio o la cara secundaria a una inflamación granulomatosa subyacente15. Las etiologías más frecuentes son la sarcoidosis y la EC16. Puede ser el signo de presentación de la EC o ser el único foco de enfermedad15,16. En estos casos debe realizarse un exhaustivo examen del sistema digestivo y un estricto control evolutivo del paciente15,17. La inflamación evoluciona de forma persistente o recidivante, es indolora y firme al tacto16. La afectación labial se denomina «queilitis granulomatosa»15 y produce un importante aumento del tamaño. En estos casos puede aparecer edema causado por la obstrucción linfática secundaria16 y generalmente se localiza sobre un labio. Cuando la afectación es importante se producen fisuras que originan una queilitis angular o media16. La granulomatosis orofacial (GOF) también se presenta como úlceras bucales lineales crónicas, dolorosas, profundas y de bordes elevados16. También puede observarse en la mucosa bucal una imagen en empedrado7 característica que produce fibrosis y adherencias residuales16.
Otras manifestaciones descritas incluyen: hiperplasia gingival, fisuras linguales, pólipos mucosos, parálisis facial, edema facial y linfadenopatía cervical16. El conjunto formado por lengua fisurada, parálisis facial y GOF se denomina síndrome de Melkersson-Rosenthal (SMR). El diagnóstico definitivo de GOF se realiza mediante estudio histológico. Se observa una inflamación granulomatosa no necrotizante. El diagnóstico diferencial incluye otras causas de edema orofacial como sarcoidosis o SMR. También deben descartarse traumatismos, angioedema, infecciones o queilitis de Miescher. Se han descrito casos aislados de aumento del tamaño labial causados por amiloidosis, quistes o síndrome de Sjögren16.
El tratamiento de la GOF ha obtenido resultados variables17. Se han utilizado corticoides intralesionales y clofazimina16. Se ha documentado que la terapia tópica mediante corticoides7 o tacrolimus puede mejorar los síntomas17. En pacientes resistentes, se han descrito respuestas a talidomida, a bolos de corticoides intravenosos16 y a corticoides sistémicos con y sin azatioprina17. Recientemente, el infliximab ha sido usado con éxito como tratamiento alternativo18. La remisión espontánea es improbable.
Crohn perianalLas manifestaciones perianales son frecuentes y pueden preceder, suceder o aparecer simultáneamente al brote de EC. En la CU, en cambio, son excepcionales. La clínica es variable: abscesos, fisuras, fístulas, fibromas, piel edematosa, úlceras, pápulas (aspecto en empedrado) y cambios en la coloración (azulado). Regresan espontáneamente hasta en un 50% y aparecen casi en la mitad de los pacientes con EC19. En un estudio realizado por Rankin et al en 569 pacientes con EC, se demostró que el 76% con ileocolitis presentaron afectación perianal y que existía una asociación positiva significativa con otras manifestaciones extraintestinales. El estudio histopatológico de estas lesiones muestra múltiples granulomas no necrotizantes típicos de EC y, a diferencia del Crohn metastásico, estos granulomas son contiguos a la enfermedad intestinal.
Las fístulas son las complicaciones más frecuentes de la EC perianal (43%)20. Son la comunicación entre dos órganos recubiertos de epitelio y representan la extensión de úlceras o fisuras desde el tubo digestivo hasta órganos adyacentes. Se clasifican según su localización: internas, externas y perianales. Mientras las primeras terminan en diversos órganos internos (genitales, vejiga urinaria, estómago), las externas desembocan en la superficie cutánea. Las fístulas perianales son muy dolorosas, recurren con frecuencia21 y disminuyen gravemente la calidad de vida del individuo. Tienen origen en la unión anorrectal y mediante la formación de abscesos entre los esfínteres anales interno y externo pueden destruir su anatomía y funcionalidad22. En los casos resistentes a terapia convencional y cirugía (proctectomía22 o fistulotomía20), se puede añadir terapia inmunosupresora con azatioprina (2,5mg/kg/d) o mercaptopurina (1,5mg/kg/d) con limitada eficacia21. Actualmente infliximab es una opción consolidada19,21 y se considera tratamiento de elección junto a la cirugía en pacientes resistentes a tratamientos médicos habituales23. En la literatura existen casos que han respondido a adalimumab24.
Las úlceras pueden ser asintomáticas y regresar espontáneamente25. En casos persistentes y sintomáticos, la terapia tópica sólo mejora los síntomas. El único procedimiento curativo es la administración de infliximab25. Si evoluciona tórpidamente puede desarrollar abscesos o fístulas secundarias25. Los abscesos se identifican fácilmente, observando una masa eritematosa dolorosa y fluctuante. Su drenaje para evitar complicaciones es el pilar de su manejo20,22. También puede añadirse ciprofloxacino o metronidazol22.
Las fisuras se localizan en la línea media y generalmente no son dolorosas, por lo que no precisan tratamiento. Las fisuras relacionadas con EC son diferentes a las demás; profundas y con los bordes despegados. Pueden asociarse a fístulas o a abscesos, situaciones que ocasionarán dolor y pueden requerir tratamiento analgésico, antiinflamatorio, antibiótico y quirúrgico. Cuando los procesos citados curan y cicatrizan, pueden producir estenosis secundarias a la fibrosis. Si son asintomáticas no requieren tratamiento, aunque los casos disfuncionales requieren dilatación quirúrgica25.
Crohn metastásico (cutáneo)La «EC metastásica» (ECM) es la presencia de granulomas no necrotizantes típicos pero no contiguos a las lesiones en el tubo digestivo26. Se denomina también «EC cutánea», al estar localizada en la piel. Es una entidad rara (menos de 100 casos descritos) y puede preceder a los síntomas digestivos hasta en un 20% de los casos. El 56% de las mujeres asociaron ECM a la EII. Se asocia a afectación colorrectal y no está correlacionado con la actividad de la EII.
La anatomía patológica muestra granulomas epitelioides y células grandes multinucleadas en la dermis y la hipodermis, sin necrosis. El diagnóstico diferencial debe incluir dermatosis granulomatosas, como tuberculosis, sarcoidosis, lepra, otras micobacterias atípicas, etc.
Se clasifica según su localización: genital, perianal y extragenital. En la ECM genital, las lesiones en el pene pueden ser desfigurantes y mutilantes. Por su parte, el 23% de las mujeres con EII tiene afectación vaginal contigua o no. Las lesiones contiguas (EC) incluyen abscesos en las glándulas de Bartholino, úlceras genitales y fístulas endovaginales. La afectación genital granulomatosa no contigua (ECM) afecta la región labial, vulvovaginal27 y perineal. Su tratamiento médico es controvertido y su eficacia es muy limitada. Se ha documentado respuesta en ECM con ciclosporina. También se han utilizado corticoides tópicos y sistémicos asociados a azatioprina, sulfasalazina y metronidazol27. Un caso con lesión vulvar fue tratado con éxito con infliximab y azatioprina27. La forma extragenital tiene predilección por extremidades inferiores y plantas. También se localiza en tronco y abdomen28 y se publicó un caso con ECM facial. Tiene tendencia a afectar las flexuras27 y puede generalizarse de forma excepcional.
Se presenta como pápulas, placas o nódulos que pueden ulcerarse. Las lesiones son firmes al tacto y tienen aspecto granulomatoso. El tratamiento médico no obedece a ninguna guía y ha obtenido resultados pobres. Consiste en la combinación individualizada de esteroides sistémicos y tópicos, antibióticos e inmunosupresores, como la azatioprina28. La cirugía está indicada como terapia de segunda línea. En los pacientes refractarios se han obtenido resultados satisfactorios con adalimumab29 y con infliximab27,30 (en monoterapia o asociado a metrotexato)28. La ECM perineal es infrecuente y puede aparecer varios años pos-proctectomía. Es más común en las mujeres que en los hombres (5:2) y la edad media se sitúa en 36 años. Se aprecian úlceras, fisuras y placas vegetantes. En este grupo se han obtenido mejores resultados con el tratamiento quirúrgico, ya que los fármacos han ofrecido pobres beneficios.
Cáncer y ECSe han descrito pacientes con carcinomas cutáneos secundarios a las lesiones inflamatorias crónicas de la EII. Aunque de forma ocasional (61 casos publicados hasta el año 2010)31, la fístula perianal representa la lesión más frecuentemente implicada, especialmente si se origina en el recto31. La patogenia no está clara. En un estudio31 que incluye los 61 casos publicados hasta la fecha, el 61% (37 pacientes) fueron mujeres y el carcinoma más frecuentemente asociado fue el adenocarcinoma (59%), seguido por el carcinoma escamoso (31%). En otro trabajo se han estudiado 6 pacientes con EC y adenocarcinoma anal asociado a fístula previa (AAAF)32. A todos se les realizó resección abdominoperineal (RAP) y 4 recibieron quimioterapia. Cuatro pacientes fallecieron con metástasis. Iesalnieks et al concluyeron que el AAAF en pacientes con EC tiene un pronóstico muy pesimista (supervivencia del 54% a los tres años pos-RAP)32. El largo tiempo de evolución de las fístulas, la inmunosupresión propia de la EEI y el empleo de fármacos inmunosupresores parecen ser los factores de riesgo en estos pacientes, que deben ser vigilados estrechamente32.
Dermatosis inespecíficasEritema nudosoEl eritema nudoso (EN) es una respuesta de hipersensibilidad retardada a una amplia variedad de estímulos antigénicos, como bacterias, virus o agentes químicos33. Es idiopático en un tercio de los casos aproximadamente y se puede asociar hasta en el 60-70% a otros procesos (tabla 3)33. En niños, el agente causal más frecuente es el estreptococo β-hemolítico del grupo A. El EN es la dermatosis más frecuente en pacientes con EII, afectando entre el 2 y 20% de estos pacientes4,6,7. Es más frecuente en mujeres entre los 20 y 40 años, lo que podría ser una consecuencia de su relación con la sarcoidosis6,33. Es más prevalente en EC que en CU6. Se correlaciona con la actividad de la EII, aunque no así con su gravedad o pronóstico6,7,33. Se ha descrito como signo de presentación de EII8.
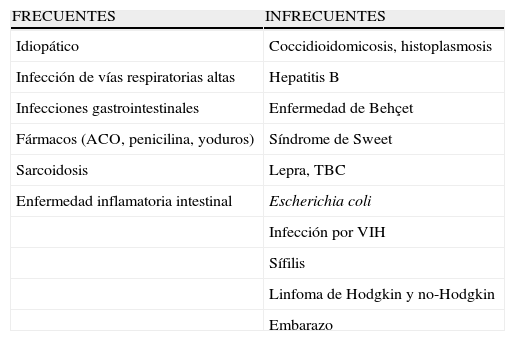
Desencadenantes del eritema nudoso. Lo más común es que no pueda relacionarse con ningún factor desencadenante (idiopático)
| FRECUENTES | INFRECUENTES |
| Idiopático | Coccidioidomicosis, histoplasmosis |
| Infección de vías respiratorias altas | Hepatitis B |
| Infecciones gastrointestinales | Enfermedad de Behçet |
| Fármacos (ACO, penicilina, yoduros) | Síndrome de Sweet |
| Sarcoidosis | Lepra, TBC |
| Enfermedad inflamatoria intestinal | Escherichia coli |
| Infección por VIH | |
| Sífilis | |
| Linfoma de Hodgkin y no-Hodgkin | |
| Embarazo |
ACO: anticonceptivos orales; CU: colitis ulcerosa.; EC: enfermedad de Crohn;
TBC: tuberculosis; VIH: virus de la inmunodeficiencia humana.
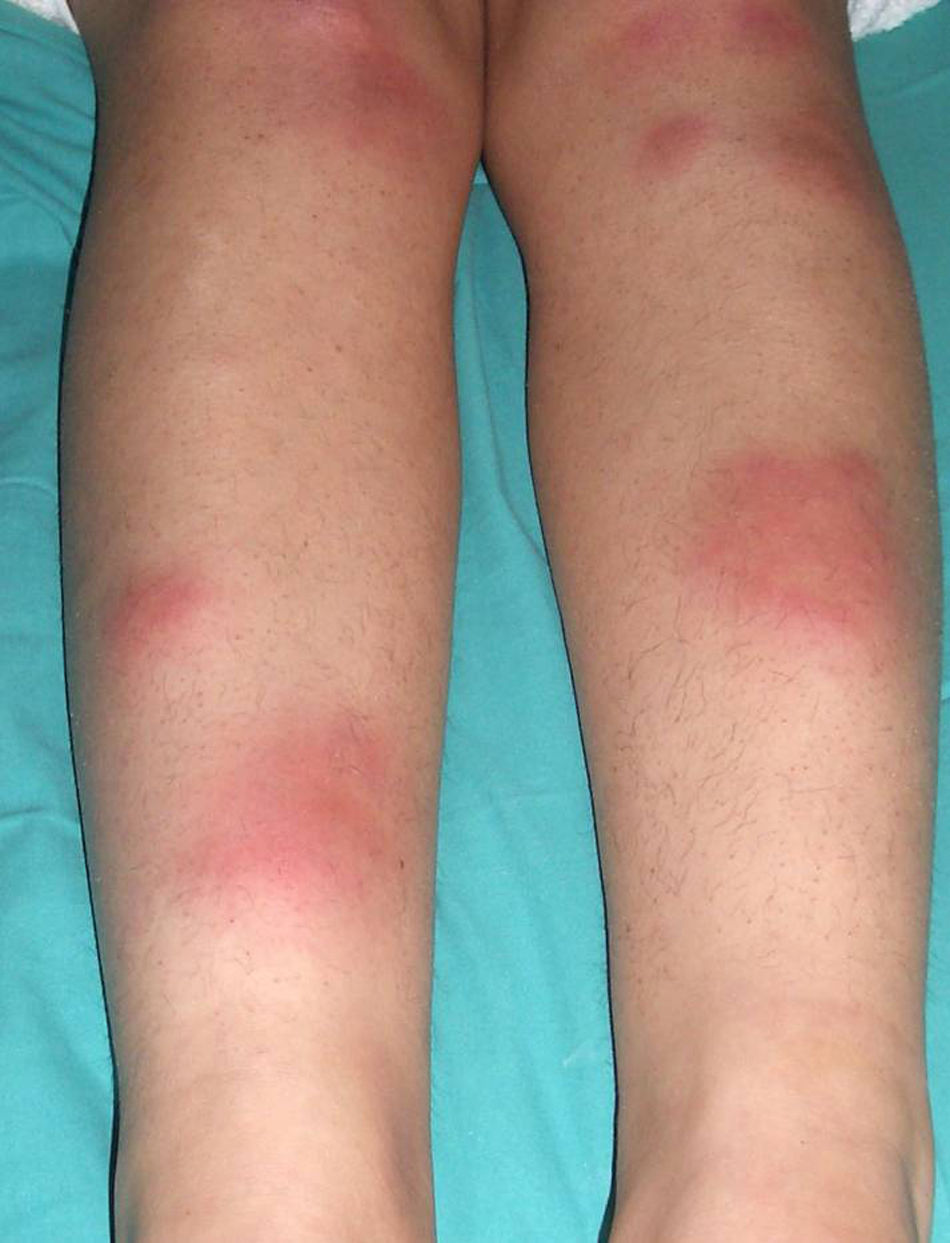
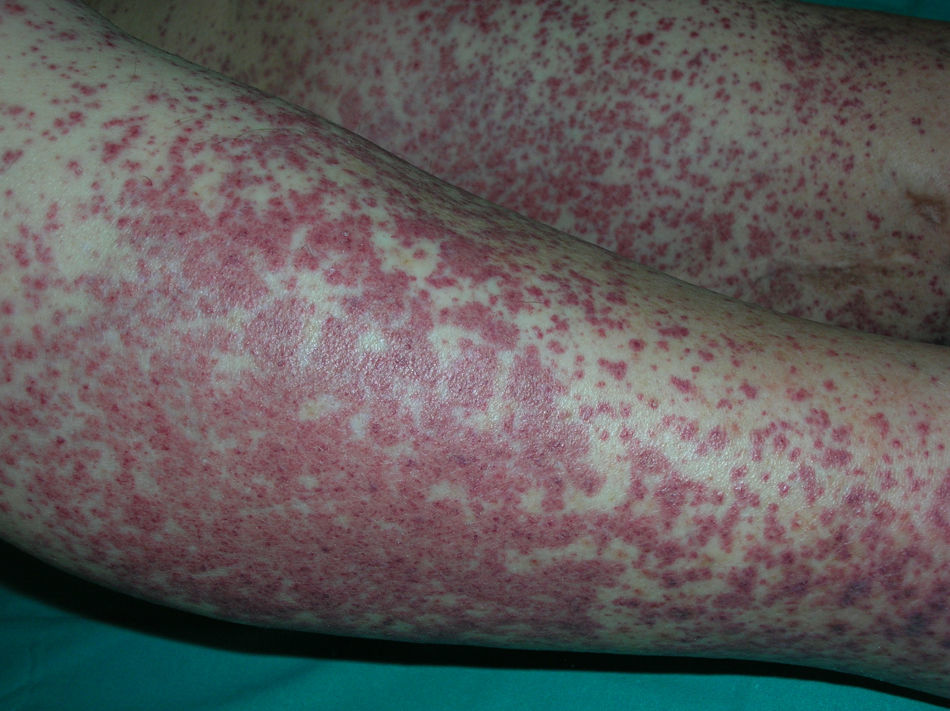
El EN se presenta en brotes caracterizados por nódulos subcutáneos inflamatorios eritematosos, calientes y dolorosos (fig. 1). Las lesiones tienen un diámetro de 1-5cm y su distribución es bilateral y simétrica. Afecta predominantemente la zona anterior de las extremidades inferiores, aunque puede aparecer en extremidades superiores, tronco o cara33. Característicamente no se ulceran y curan en unas 6 semanas33. En ocasiones se acompaña de fiebre, artritis y sinovitis.

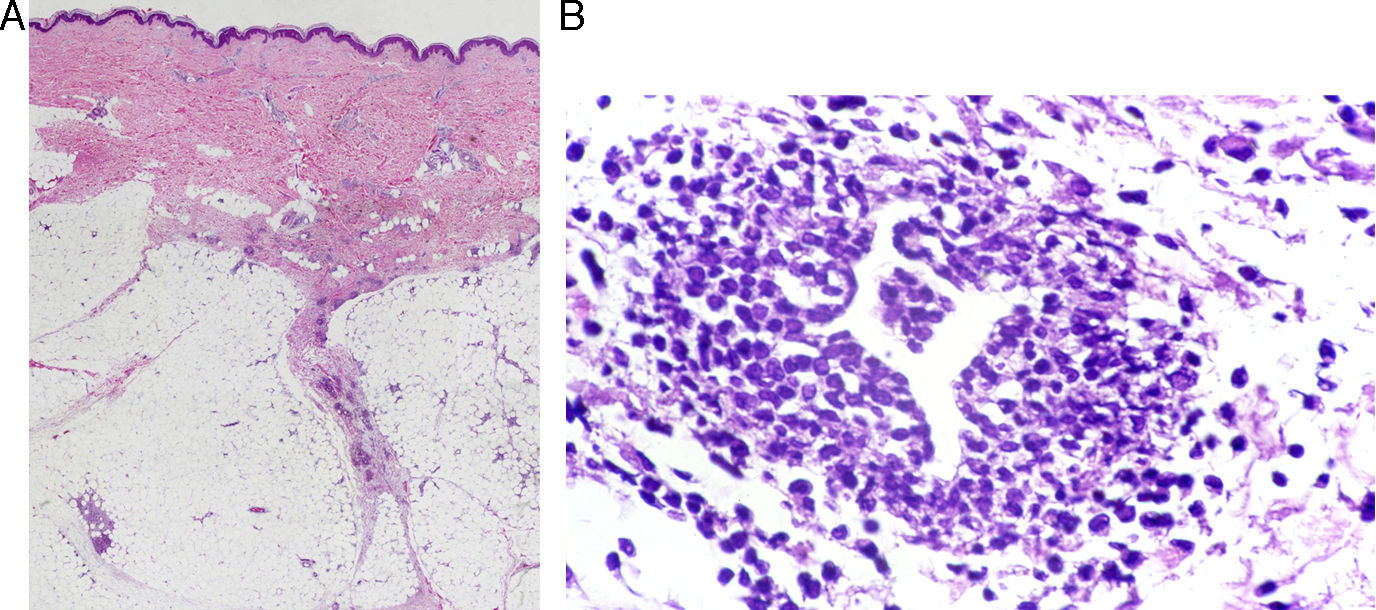
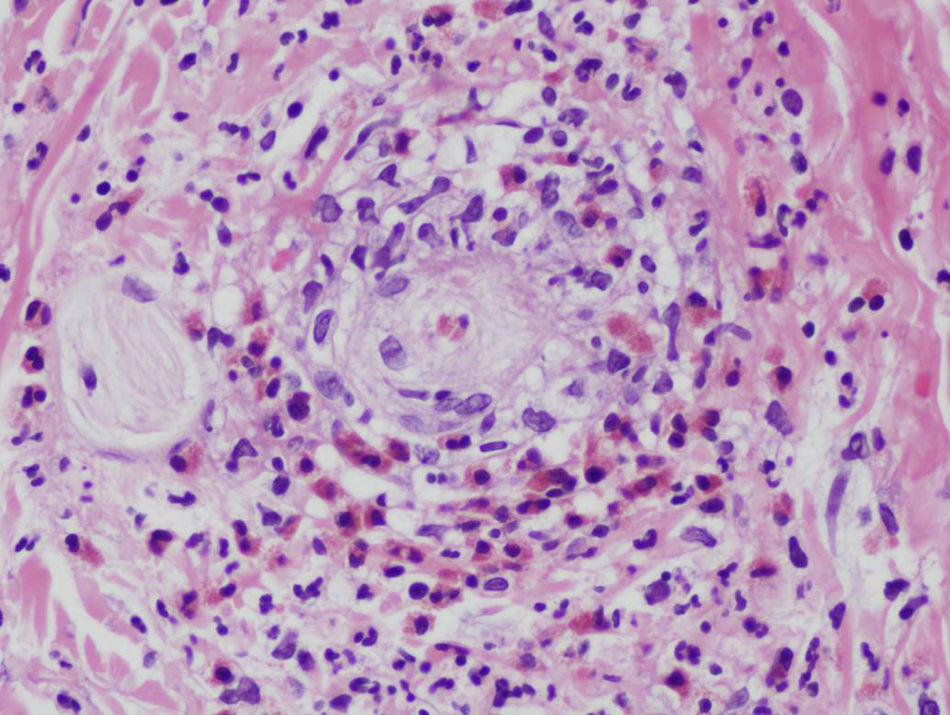
El diagnóstico definitivo se realiza mediante biopsia incisional profunda. El estudio histopatológico muestra una paniculitis de predominio septal sin vasculitis (fig. 2A)34. Las características del infiltrado varían según la fase de la lesión, predominando inicialmente los neutrófilos con edema y hemorragia. La lesión puede extenderse hasta el tejido periseptal de los lóbulos y adoptar un predominio lobulillar. Sin embargo, en contra de lo que sucede en una paniculitis lobulillar, no se acompaña de necrosis grasa significativa. En lesiones más avanzadas domina el componente de linfocitos y células gigantes multinucleadas, acompañado de fibrosis. Puede apreciarse un infiltrado perivascular linfocitario en la dermis superficial, media y profunda34. Es muy característica la formación de granulomas de Miescher (fig. 2B) dentro de los septos, compuestos por colecciones de macrófagos alrededor de neutrófilos o de espacios de tipo hendidura o tabique34. Cuando la lesión progresa, los macrófagos son sustituidos por células gigantes multinucleadas y epitelioides. A pesar de que existe importante fibrosis septal en estadios finales, la lesión cura sin dejar cicatriz34. Debido al componente granulomatoso que existe en lesiones tardías de EN, puede estar indicado descartar una sarcoidosis. Aunque el diagnóstico suele ser claro, deben considerarse otros tipos de paniculitis cuando las lesiones son atípicas, afectan localizaciones inusuales o permanecen más de 6 semanas.
 El eritema nudoso es la paniculitis de predominio septal más frecuente y no se acompaña de vasculitis. B) Granuloma de Miescher. (Por cortesía de Santos-Briz A, MD).")
El pronóstico es excelente y las recidivas son raras. En muchos casos regresa espontáneamente y medidas físicas, como compresión, elevación del miembro y reposo pueden ser suficientes. También se usan antiinflamatorios no esteroideos, prednisona, yoduro potásico, colchicina e hidroxicloroquina. En casos de EN refractario, se ha descrito respuesta a adalimumab estando asociado a EC o no35.
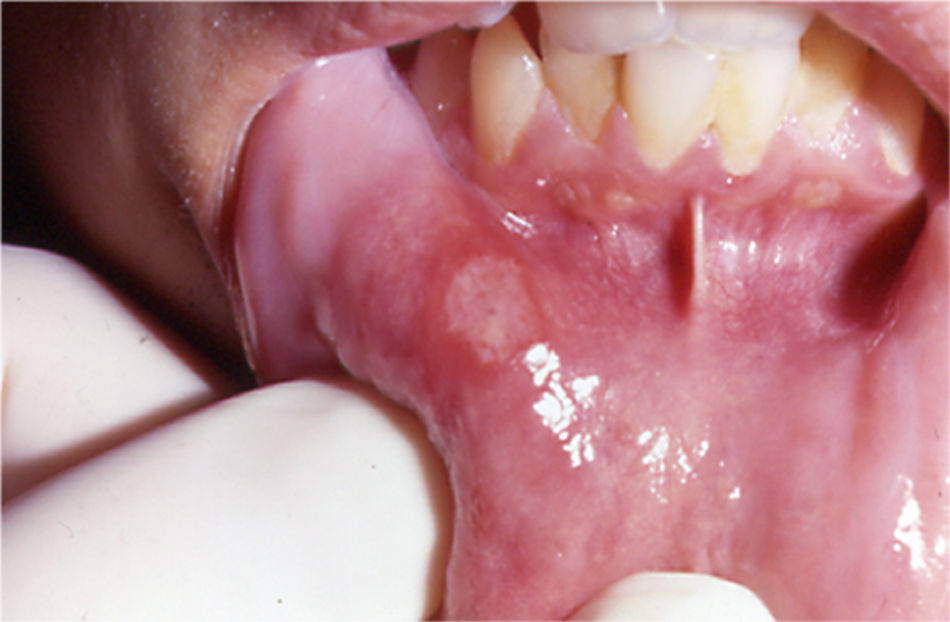
Estomatitis aftosa recidivanteEn general, las manifestaciones orales se describen en el 4-16% de los pacientes con EII y pueden ser el signo de presentación hasta en un 60% de los casos17. La estomatitis aftosa recidivante (EAR) aparece en el 8 y el 6% de los pacientes con CU y EC, respectivamente33. Además, puede estar asociada, entre otros, a los siguientes procesos sistémicos: enfermedad de Behçet, síndrome de Reiter, enteropatía sensible al gluten, deficiencias vitamínicas (B12 y ácido fólico) e inmunodeficiencias tales como la neutropenia cíclica y el VIH7,33. Clínicamente pueden ser indistinguibles7.
Su patogenia es desconocida, aunque se piensa que su origen es multifactorial. Las aftas son dolorosas e interfieren en las actividades básicas de la vida diaria, como comer o hablar33. Tienden a localizarse en el suelo de la boca, encías, lengua, labios, úvula y paladar33. Clínicamente, la EAR tiene tres presentaciones. La forma menor33 es la más frecuente y consiste en la presencia de úlceras superficiales, dolorosas, de color blanco cremoso y rodeadas de un halo eritematoso (fig. 3). Son menores de 5mm, redondas u ovales y no suelen dejar cicatriz. La forma mayor33 puede ir acompañada de fiebre y se caracteriza por úlceras de hasta 3cm, profundas, más persistentes y que pueden dejar cicatriz residual. También existe una forma herpetiforme, muy infrecuente33. Los casos asociados a EII suelen pertenecer a las formas menor o mayor33.

Histopatológicamente no se aprecian datos específicos y debe establecerse la correlación con la clínica. En ocasiones se demuestran granulomas típicos de EC. La aplicación precoz de corticoides tópicos potentes puede aliviar el dolor y acelerar la cicatrización7. Se añadirán analgésicos o anestésicos tópicos (lidocaína al 2%)7 si procede. Debe reservarse el uso de dapsona, colchicina y talidomida para casos refractarios33. Siempre debe tratarse la enfermedad de base7.
Dermatosis neutrofílicasPioderma gangrenosoEl pioderma gangrenoso (PG) es una enfermedad cutánea ulcerosa, crónica y recurrente36. Es discretamente más frecuente en mujeres entre los 20 y 50 años de edad37. En el 25-50% de los casos es idiopático. El resto (50-75%) se asocia a patologías sistémicas, como EII (CU y EC), artritis (artritis reumatoide, seronegativas, espondilitis de la EII), trastornos hematológicos (el más frecuente es la gammapatía monoclonal tipo IgA, pero también tricoleucemia o mielofibrosis), vasculitis y otras dermatosis neutrofílicas (enfermedad de Behçet, dermatosis pustulosa subcorneal y síndrome de Sweet)37,38. También se relaciona con diversos procesos cardíacos y pulmonares.
El PG es la segunda dermatosis más frecuente en pacientes con EII (afecta al 2-12%), siendo descrito en el 1-10% de pacientes con CU y en el 0,5-20% con EC38. Por otra parte, aproximadamente un tercio de los pacientes con PG presentan EII38. A pesar de que la patogenia es desconocida, se piensa en la existencia de un mecanismo inmunológico subyacente, como la disfunción de la inmunidad celular y humoral37. Apoya esta hipótesis la aparición del fenómeno de patergia hasta en el 50% de los casos33. También se han descrito casos de PG familiar, como en el síndrome PAPA, con herencia autonómica dominante con afectación del cromosoma 15q. Este síndrome incluye artritis purulenta estéril, acné y PG37. No es infrecuente la aparición de PG secundario al yoduro potásico, ya que puede actuar como factor estimulante de colonias granulocíticas. También se ha documentado durante el tratamiento con isotretinoína por vía oral.
En general, la evolución clínica puede seguir dos patrones: agudo y agresivo o crónico e indolente37. En el primero aparecen de forma abrupta numerosas lesiones dolorosas que se acompañan de sintomatología sistémica y fiebre. Las lesiones se generalizan, progresan rápidamente y tiene peor pronóstico. La evolución lenta típica del segundo tipo permite la formación de tejido de granulación y la regresión espontánea en algunos casos, por lo que el pronóstico mejora drásticamente37.
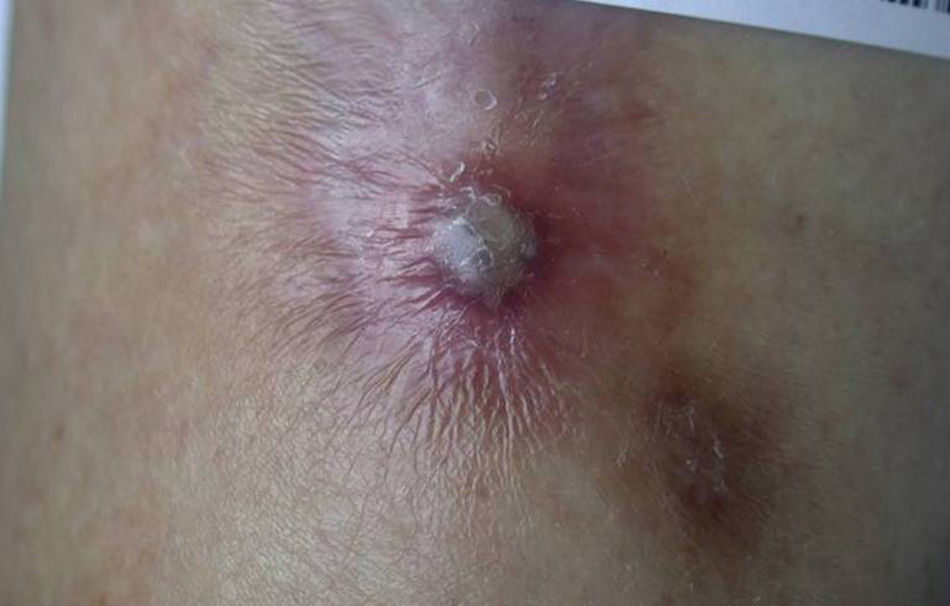
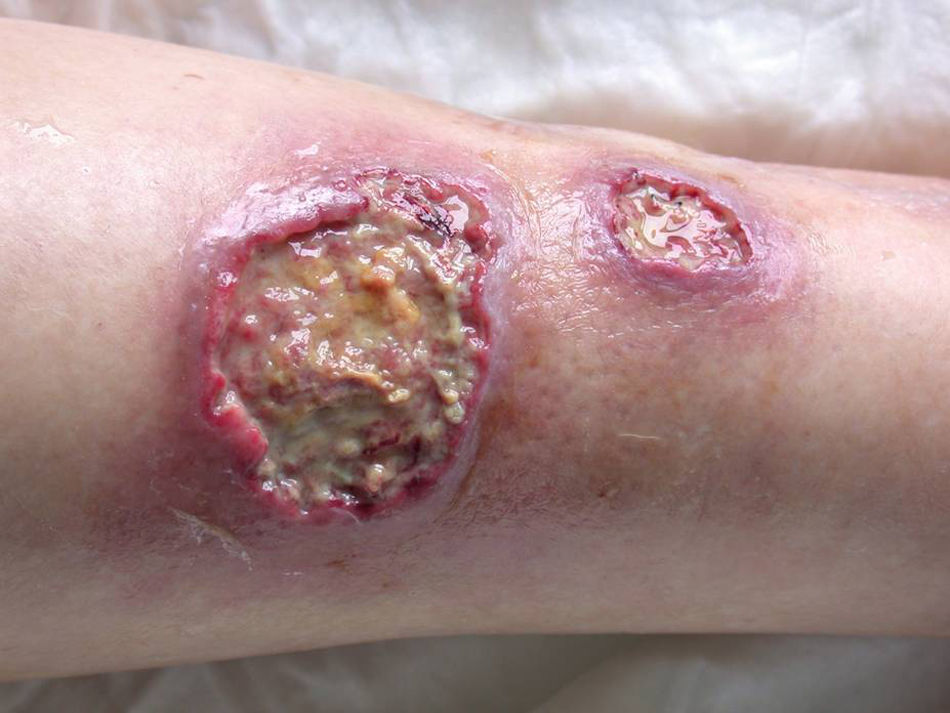
Existen cuatro grandes grupos de PG: clásico, pustuloso, ampollar y vegetante7. Durante su evolución, la forma clásica tiene distintas fases clínico-histológicas y pronósticas37. Inicialmente, aparecen papulopústulas foliculares, estériles y dolorosas sobre una base eritematosa indurada o nodular (fig. 4)37. Hasta en el 70% se localiza en las extremidades inferiores5, aunque puede aparecer en cualquier localización como mucosas37 y tejidos periestomales33. La lesión resultante es una úlcera profunda, necrótica y purulenta (no infecciosa), con bordes activos que cronifican la lesión y se extienden centrífugamente (fig. 5). Las úlceras superficiales afectan la dermis y las profundas hasta el tejido graso subcutáneo o fascia37. La reepitelización se produce de forma inversa, desde los bordes hacia el centro, dejando cicatriz atrófica e hiperpigmentación residual37. Mientras esto ocurre, la enfermedad puede seguir activa en otras regiones, apareciendo nuevas lesiones que pueden confluir formando úlceras arciformes37. La forma pustulosa está asociada a procesos linfoproliferativos, EII, síndrome de artritis-dermatosis Asociado al intestino y enfermedad de Behçet. Se desarrollan pústulas estériles, foliculares y dolorosas que no progresan hacia la formación de úlceras. La forma atípica o ampollar está relacionada con fármacos y trastornos mieloproliferativos como el síndrome mielodisplásico y la leucemia mieloide aguda. Debe realizarse el diagnóstico diferencial (DD) con la variante ampollar del síndrome de Sweet. El PG vegetante37 o granulomatoso superficial tiene un comportamiento menos agresivo que los anteriores y no se asocia a procesos sistémicos. Se forman úlceras de crecimiento lento, superficiales, verrugosas y no purulentas.


Según Ruocco et al, actualmente en la clasificación del PG se incluyen otras formas37: periestomal, genital, infantil y extracutáneo. En el primer grupo, el fenómeno de patergia parece tener un papel fundamental debido al contacto con el estoma, heces y adhesivos. Las úlceras del PG genital difieren clínicamente del clásico localizado en esa región y debe descartarse enfermedad de Behçet. Entre el 3 y el 4% de casos totales de PG se desarrollan durante la niñez37 y muestran mayor predilección por la región anal y genital. El pronóstico es favorable. A nivel extracutáneo, se han descrito presencia de infiltrados neutrofílicos estériles en pulmón (órgano más frecuente), corazón, sistema nervioso central, ganglios linfáticos, tracto gastrointestinal, hígado, bazo u ojo38.
El estudio histopatológico en el PG es inespecífico5. Se observa edema y un denso infiltrado neutrofílico dérmico en muestras tomadas de bordes activos y necrosis con células mononucleadas acompañantes en las biopsias de zonas centrales33. Es imprescindible excluir otras entidades causantes de úlceras y llevar a cabo una correcta correlación clínico-histológica debido a la carencia de hallazgos específicos por separado36.
No existen unos criterios específicos de tratamiento37. En los casos asociados a otras enfermedades, como EII, el tratamiento de éstas puede mejorar las lesiones de PG37. Deben minimizarse los traumatismos quirúrgicos (patergia). En los casos leves y localizados39 está indicada la terapia tópica y medidas como la compresión de la zona, el reposo y la elevación del miembro afectado pueden ser de utilidad. La inyección en los bordes de la úlcera de 5mg/ml de acetato de triamcinolona dos veces por semana es el tratamiento más efectivo37. Ha mostrado beneficios la inyección de ciclosporina intralesional. También pueden usarse tacrolimus 0,1%, ácido 5-aminosalicílico y corticoides de potencia muy alta37.
En la literatura existe una amplia lista de fármacos sistémicos que han demostrado beneficios en los casos de mayor gravedad. Dosis altas (100-200mg/d) de prednisona o ciclos de metilprednisolona en bolos (1g/d) durante 5 días pueden ser necesarios de inicio. La sulfapiridina, la dapsona y la ciclosporina controlan y mejoran las lesiones en un número elevado de casos39. En la literatura aparecen como alternativas: talidomida, clorambucilo, azatioprina, metrotexato, antibióticos, micofenolato de mofetilo, 6-mercaptopurina y ciclofosfamida. Se han documentado buenos resultados con plasmaféresis39 e inmunoglobulinas intravenosas (IGIV)40.
Se han llevado a cabo estudios para comprobar el funcionamiento de fármacos anti factor de necrosis tumoral-α (anti-TNFα) en el PG refractario41, observándose un inicio de acción rápido y una mejoría muy llamativa. El trabajo más relevante ha sido realizado por Brooklyn et al42, en el que se demostró la superioridad de infliximab sobre placebo en PG asociado o no a EII. Otras publicaciones también muestran beneficios con infliximab43. Recientemente, adalimumab y alefacetp, una proteína recombinante que inhibe la interacción entre LFA-3 y CD2 al interferir en la activación de los linfocitos T, también han sido beneficiosos en casos de PG resistente a la terapia inmunosupresora habitual24,44.
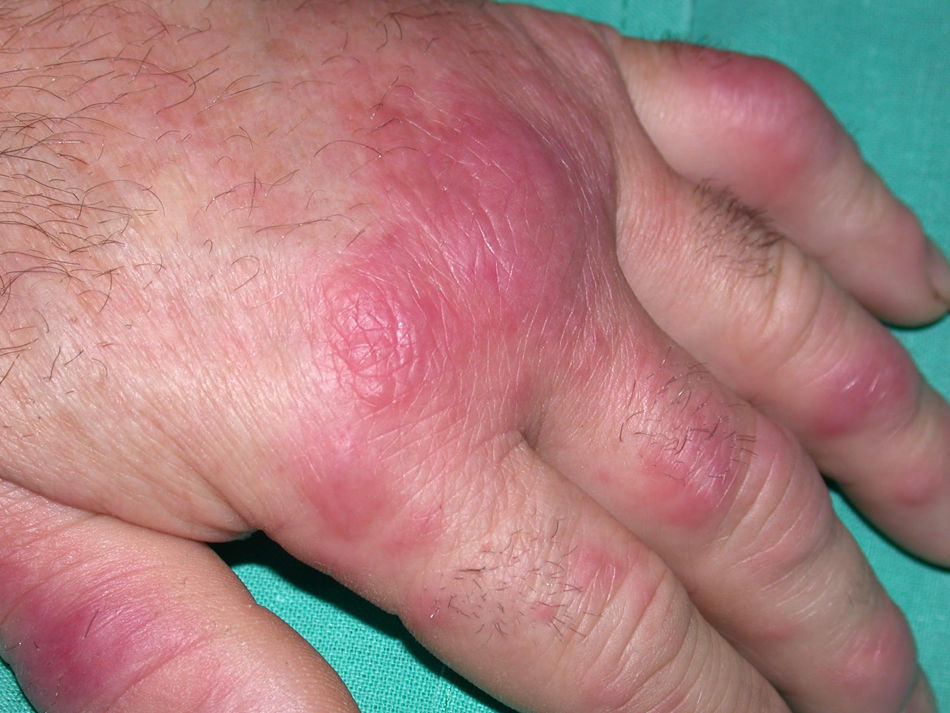
Síndrome de SweetEl síndrome de Sweet (SS) está compuesto por las siguientes manifestaciones: fiebre, neutrofilia en sangre periférica y lesiones cutáneas eritematosas y dolorosas45,46. La forma clásica es más frecuente en mujeres de edad entre 30 y 50 años y no presenta predilección racial33,45. Tiene una distribución mundial y es poco común47. La patogenia es desconocida45. Podría ser consecuencia de una reacción inmune de tipo celular linfocitoT-dependiente desencadenada por un antígeno, proceso que aumentaría la producción de ciertas citocinas, como el factor de necrosis tumoral-α (TNF-α), interferón-γ (INF-γ) e interleucina-1 (IL-1)48. El SS puede ser idiopático o asociarse a procesos malignos (donde las recidivas son más frecuentes)49–51, infecciones (los focos más frecuentes son las vías respiratorias superiores y el abdominal48), enfermedad tiroidea, embarazo45 y trastornos autoinmunitarios e inflamatorios (EII, sarcoidosis, enfermedad de Behçet)52–54.
Se han documentado pacientes con SS y EII en los que se ha observado la coexistencia de PG55 o EN. Esta relación puede ser debida al carácter reactivo y al papel fundamental desarrollado por los neutrófilos en la patogenia de estas dos entidades48. Los fármacos pueden actuar como desencadenantes y el más frecuente es el factor de estimulación de colonias de granulocitos (G-CSF)56,57. En el 12% de los casos de SS se objetiva enfermedad sistémica subyacente, siendo las más frecuentes la EC y la CU33. El 87% de los casos son mujeres y es más frecuente cuando se acompaña de otras manifestaciones extraintestinales (77%)54. Debido a que todos los pacientes comunicados han presentado afectación colónica, existen hipótesis que relacionan la presencia de bacterias o antígenos en el colon con la aparición de SS. Sólo en el 20% de los casos existió patología ileal acompañante54. Aunque suele aparecer después (52%), también puede debutar antes (20%) o junto al brote (28%) de EII54.
El cuadro clínico se caracteriza por fiebre, recuento de neutrófilos elevado y aparición de pápulas, placas o nódulos dolorosos y eritematosos (fig. 6)46. Debido al denso edema dérmico subyacente, las lesiones adoptan un aspecto transparente y brillante que forman pseudovesículas y pseudopústulas sólidas al tacto45. Puede acompañarse de artromialgias, cefalea y conjuntivitis. Se localiza preferentemente en cabeza, cuello y extremidades. El fenómeno de patergia puede estar presente. Se han comunicado casos fotodistribuidos o con lesiones localizadas sobre linfedema crónico posmastectomía en el brazo ipsilateral57.

Aparecen recurrencias en un 25-50% de los casos, pueden regresar espontáneamente y generalmente curan sin dejar cicatriz33. La presencia de nódulos subcutáneos eritematosos y dolorosos es más frecuente en extremidades y se denomina «SS subcutáneo»58, proceso ya mencionado por Sweet en su artículo original46. Ocasionalmente el SS se presenta mediante pústulas estériles localizadas en la cara dorsal de las manos59,60. Esta variante fue incluida en 1988 en un grupo denominado «vasculitis pustulosas» debido a la presencia de vasculitis leucocitoclástica en numerosos casos. En un 40% de los pacientes se acompañaba de lesiones en otras localizaciones45 y también ha sido relacionada con la colitis ulcerosa. Además, se ha descrito afectación extracutánea del SS (tabla 4)47,61. Las lesiones en mucosas son más frecuentes en procesos hematológicos45,49. La asociación entre SS y leucemia cutis ha sido recientemente establecida45. Podría ser un proceso paraneoplásico, así como ser inducido por tratamientos (ácido all-trans retinoico, G-CSF56) o ser una dermatosis coincidente45.
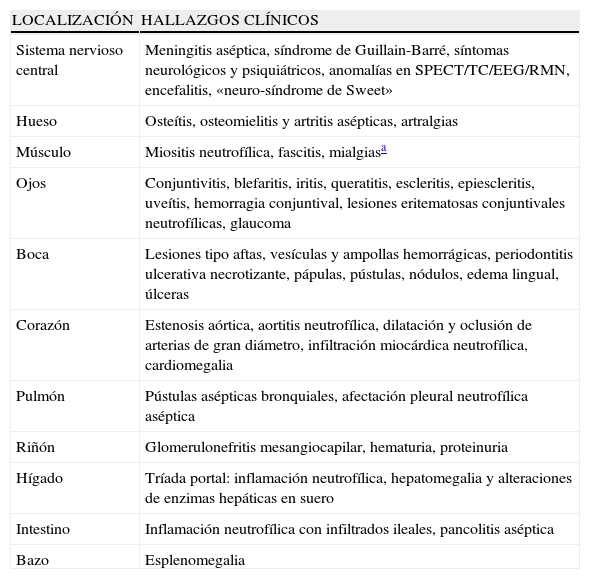
Manifestaciones extracutáneas en el síndrome de Sweet (SS). Se denomina «neuro-SS» a la presencia de una encefalitis benigna recurrente acompañada de una dermatosis neutrofílica
| LOCALIZACIÓN | HALLAZGOS CLÍNICOS |
| Sistema nervioso central | Meningitis aséptica, síndrome de Guillain-Barré, síntomas neurológicos y psiquiátricos, anomalías en SPECT/TC/EEG/RMN, encefalitis, «neuro-síndrome de Sweet» |
| Hueso | Osteítis, osteomielitis y artritis asépticas, artralgias |
| Músculo | Miositis neutrofílica, fascitis, mialgiasa |
| Ojos | Conjuntivitis, blefaritis, iritis, queratitis, escleritis, epiescleritis, uveítis, hemorragia conjuntival, lesiones eritematosas conjuntivales neutrofílicas, glaucoma |
| Boca | Lesiones tipo aftas, vesículas y ampollas hemorrágicas, periodontitis ulcerativa necrotizante, pápulas, pústulas, nódulos, edema lingual, úlceras |
| Corazón | Estenosis aórtica, aortitis neutrofílica, dilatación y oclusión de arterias de gran diámetro, infiltración miocárdica neutrofílica, cardiomegalia |
| Pulmón | Pústulas asépticas bronquiales, afectación pleural neutrofílica aséptica |
| Riñón | Glomerulonefritis mesangiocapilar, hematuria, proteinuria |
| Hígado | Tríada portal: inflamación neutrofílica, hepatomegalia y alteraciones de enzimas hepáticas en suero |
| Intestino | Inflamación neutrofílica con infiltrados ileales, pancolitis aséptica |
| Bazo | Esplenomegalia |
Modificado de: Cohen and Kurzroc. Int J Dermatol 2003; 42: 761–778.
EEG: electroencefalograma; RMN: resonancia magnética nuclear; SPECT: tomografía computarizada por emisión de fotones individuales; TC: tomografía computarizada.
El estudio histológico demuestra edema en dermis papilar, junto con un denso infiltrado inflamatorio superficial compuesto predominantemente por neutrófilos maduros, con un número variable de linfocitos o macrófagos. Se han descrito casos idiopáticos e inducidos por fármacos con presencia de eosinófilos. El infiltrado puede ser difuso o bien puede adoptar una morfología nodular o perivascular62. Es frecuente la presencia de leucocitoclasia (fragmentación nuclear de los neutrófilos) y los vasos de pequeño tamaño muestran dilatación y tumefacción endotelial. En un estudio de 31 casos de SS realizado por Ratzinger et al se observó la presencia de cambios vasculíticos en 23 pacientes (74%)62. La presencia de una verdadera vasculitis leucocitoclástica es controvertida, llevando a ciertos autores a plantear la posibilidad de tratarse de un epifenómeno60. La epidermis suele estar respetada. Ocasionalmente puede observarse exocitosis de neutrófilos. Cuando las células inflamatorias migran al tejido graso subcutáneo se desarrolla una paniculitis neutrofílica denominada «SS subcutáneo», que puede ser lobulillar, septal o mixta58,63. En estos casos se puede afectar el tejido adiposo solamente o incluir también la dermis62,63. Debe realizarse el diagnóstico diferencial con el EN. Requena et al definieron como «SS histiocitoide» al infiltrado dérmico compuesto por células histiocito-like que en realidad eran células mieloides inmaduras64.
El tratamiento de elección son los corticoides sistémicos a dosis de 30-60mg/d, siendo la respuesta en general satisfactoria39. También se usan ciclos de metilprednisolona en bolos (1g/d) durante 3-5 días. Está indicada la terapia tópica mediante el uso de corticoides tópicos39 de muy alta potencia (propionato de clobetasol al 0,05%)33 o corticoides intralesionales (acetato de triamcinolona a dosis entre 3-10mg/ml)45. El yoduro potásico y la colchicina son otros tratamientos de primera línea33,39. Indometacina, clofazimina, ciclosporina y dapsona son fármacos alternativos39. Se ha administrado infliximab con éxito en un paciente con enfermedad de Crohn que también presentaba síndrome de Sjögren y SS65. Los casos extracutáneos suelen responder a la misma terapia.
Síndrome dermatosis-artritis asociado al intestinoProceso que se manifiesta en pacientes sometidos a cirugía de derivación intestinal a consecuencia de la formación de un asa ciega66. En el interior del bucle se produce un sobrecrecimiento bacteriano que libera antígenos a la circulación, los cuales forman complejos inmunitarios que se depositan en el tejido sinovial y en la piel67. También se ha demostrado quimiotaxis de neutrófilos67. Las bacterias más frecuentemente implicadas son Escherichia coli, Bacterioides fragilis y algunas especies de estreptococos. La remisión del cuadro con tratamiento antibiótico apoya esta hipótesis. Los casos asociados a EII son más frecuentes en CU debido al mayor número de intervenciones quirúrgicas que se realizan en esta entidad.
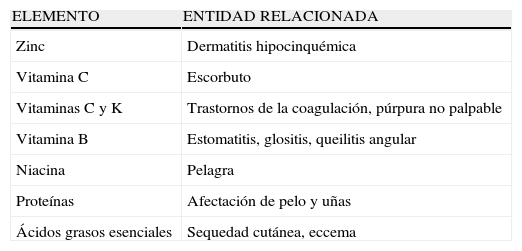
La clínica puede aparecer después de 1-6 años después de la cirugía y se caracteriza por lesiones cutáneas y artritis, consecuencia del depósito de inmunocomplejos en estos tejidos. Este cuadro puede ir precedido de síntomas constitucionales similares a la enfermedad del suero como artromialgias, fiebre y malestar general. En la piel se forman pápulas eritematosas que evolucionan a vesiculopústulas de 2-4mm de diámetro66. Sus localizaciones electivas son el tronco superior y la parte proximal de las extremidades. Su duración oscila entre 2 y 6 días y puede recurrir entre las 2 y 6 semanas en forma de brotes66. Los síntomas articulares corresponden a una poliartritis migratoria no destructiva, que puede acompañarse de tenosinovitis. Existen complicaciones sistémicas como insuficiencia hepática, litiasis renal por oxalato cálcico y litiasis biliar. A causa de la diarrea persistente, secundaria a este proceso y a la propia actividad de la EII, se desarrollan trastornos derivados de la malabsorción (tabla 5).
Dermatosis relacionadas a malabsorción intestinal
| ELEMENTO | ENTIDAD RELACIONADA |
| Zinc | Dermatitis hipocinquémica |
| Vitamina C | Escorbuto |
| Vitaminas C y K | Trastornos de la coagulación, púrpura no palpable |
| Vitamina B | Estomatitis, glositis, queilitis angular |
| Niacina | Pelagra |
| Proteínas | Afectación de pelo y uñas |
| Ácidos grasos esenciales | Sequedad cutánea, eccema |
Debido a sus características clínicas y topográficas, la dermatitis hipocinquémica debe ser diferenciada de la enfermedad de Crohn perianal y metastásica mediante estudio histológico
Algunos casos pueden acompañarse de EN o paniculitis nodular no supurativa. Se han descrito erupciones vesiculopustulares idénticas a la descrita sin estar asociadas a cirugía previa. Estos casos no se acompañan de artropatía y están relacionados a patología intestinal subyacente67, hecho que indica que la estasis intestinal podría tener un papel patogénico importante en el crecimiento bacteriano. Algunos autores sugieren que representa una forma abortiva de PG68 y aparecen con cierta frecuencia en la CU68.
Histopatológicamente se observa un infiltrado perivascular y nodular dérmico formado predominantemente por neutrófilos y que puede extenderse hasta la epidermis o el panículo. Es posible que se aprecie leucocitoclasia y edema papilar y reticular. Aunque se observan con frecuencia signos de vasculopatía sin vasculitis, algunos autores incluyen esta entidad en la vasculitis pustulosas67. En los casos asociados a EII (mayormente CU) debe tratarse la enfermedad de base. La reintervención del paciente resecando el bucle ciego es curativa66. El tratamiento médico incluye antibióticos sistémicos como tetraciclinas, clindamicina y metronidazol66, entre otros. Los corticoides sistémicos consiguen una mejoría sintomática. Se ha documentado un caso que respondió adecuadamente a micofenolato de mofetilo.
Dermatosis neutrofílica IgA intraepidérmicaProceso autoinmunitario que actualmente se considera, junto a la dermatosis pustulosa subcorneal, un subtipo de pénfigo vulgar IgA69. Suele debutar en personas de mediana edad o mayores. Se han descrito casos en pacientes con CU69. Se presenta como una erupción formada por vesículas flácidas o pústulas sobre piel eritematosa, que tienden a confluir adoptando una morfología anular o «en girasol»69. Aparece con más frecuencia en pliegues axilares e inguinales, aunque también puede hacerlo en otras localizaciones70. La afectación mucosa es rara y el principal síntoma es el intenso prurito70. Existen abundantes neutrófilos junto a depósitos de IgA en las superficies celulares de los queratinocitos epidérmicos69–71, que pueden detectarse por IF directa en todos los casos y por IF indirecta en aquéllos con autoanticuerpos IgA circulantes72. El tratamiento de elección es la dapsona72. También se han administrado ciclosporina A y corticoides sistémicos con éxito69.
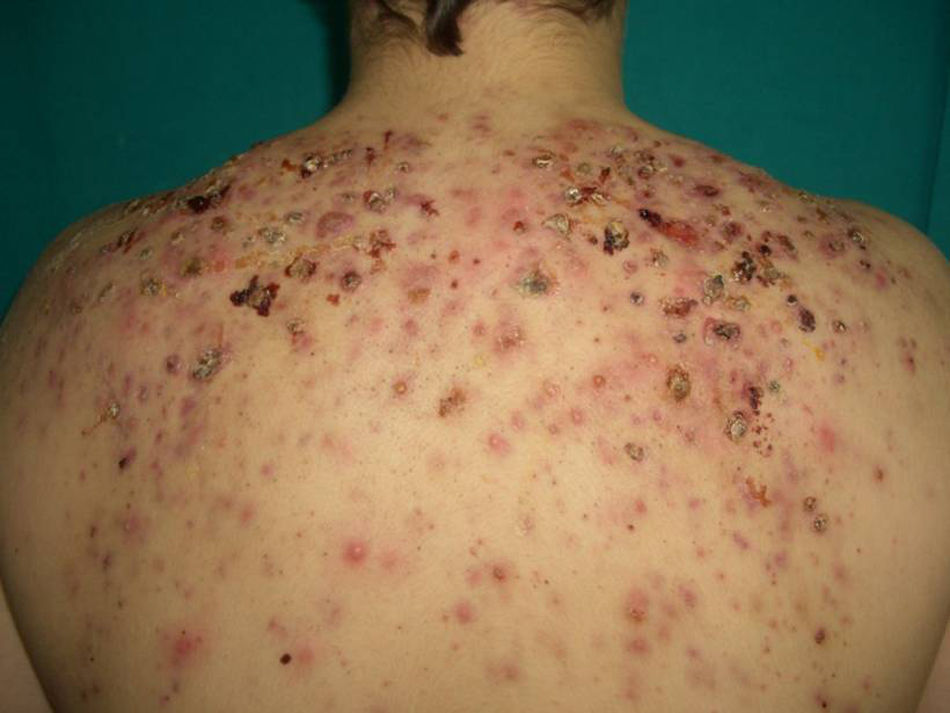
Acné fulminansEs la forma más grave de acné quístico y afecta principalmente a jóvenes entre 13 y 16 años73. Es común que los pacientes presenten acné leve o moderado previo al brote. Aunque la patogenia es desconocida, podría tratarse de un proceso mediado inmunológicamente. Paradójicamente, los pacientes con acné grave quístico pueden presentar acné fulminans (AF) causado por la propia isotretinoína73. Se inicia bruscamente con la aparición de microcomedones que confluyen formando placas inflamatorias y dolorosas y se acompaña de afectación sistémica como fiebre, leucocitosis, artromialgias y hepatoesplenomegalia (fig. 7)73. Finalmente se forman nódulos supurativos con costras serohemorrágicas, que se ulceran dejando importantes cicatrices residuales. Las zonas más implicadas son cara, cuello, pecho, espalda y brazos. Es posible que aparezcan lesiones osteolíticas en el esternón y las clavículas, entre otros huesos. También se ha descrito EN acompañante73. Su relación con EC74 ha sido documentada.

Hay autores que sugieren que el AF pertenece al síndrome sinovitis-acné-pustulosis-hiperostosis-osteítis (SAPHO)75,76. La base del tratamiento son los corticoides: tópicos, intralesionales y sistémicos. Se asocian isotretinoína y antibióticos sistémicos. En los casos secundarios a isotretinoína se puede prevenir mediante la coadministración de corticoides sistémicos junto a bajas dosis del retinoide durante el primer mes de tratamiento. Cuando en casos de AF también aparece EN, se debe asociar dapsona73. Se ha publicado un caso de AF recalcitrante que respondió a infliximab76.
VasculitisArteritis de TakayasuLa arteritis de Takayasu (AT) es una panarteritis de grandes vasos granulomatosa, crónica, recurrente y poco frecuente. También se denomina «síndrome del cayado aórtico», porque produce aneurismas y estenosis en la aorta y sus ramas, así como en las arterias coronarias y pulmonares77. Pese a relacionarse con entidades autoinmunitarias, su patogenia actualmente no se conoce. En la literatura hay 29 casos descritos de EC con AT. Además, en el 88% de los casos relacionados el diagnóstico de AT fue simultáneo o posterior78 al de EC, lo que refuerza la hipótesis que incluye la AT dentro de las manifestaciones extraintestinales de la EC77. Por otra parte, existen numerosas comunicaciones de pacientes con CU que desarrollan AT79, incluso después de estar proctocolectomizados.
La clínica se divide en dos etapas: una inicial con pulso y una tardía sin pulso. El inicio es insidioso, con síntomas constitucionales. Posteriormente aumenta la tensión arterial en las extremidades hasta desaparecer el pulso y pueden aparecer soplos, asimetría de pulsos, claudicación y signos isquémicos (incluso ceguera)77. Se han descrito alteraciones dermatológicas en el 15-20% de los pacientes. Comienza con nódulos similares al EN o al eritema indurado de Bazin y progresa en la fase tardía, simulando lesiones de vasculitis de pequeño vaso o de PG. El diagnóstico se realiza por la correlación entre la clínica, las pruebas de imagen (angiografía) y el estudio histológico.
Al microscopio, durante la fase inicial con pulso, se pueden observar signos de vasculitis granulomatosa que afecta vasos de gran tamaño de forma parcheada. El infiltrado inflamatorio está compuesto por linfocitos y células plasmáticas con cantidad variable de histiocitos y eosinófilos. En la fase tardía se aprecia una esclerosis transmural, con un ínfimo infiltrado inflamatorio. En ciertos casos se ha observado la existencia de trombosis secundarias.
La prednisona (durante 6 meses o hasta 12 años, según la respuesta) es el pilar del tratamiento. Empezar la terapia en la fase inicial es muy importante. La tasa de recidivas se reduce significativamente al asociarse ciclofosfamida. En los últimos años se han utilizado fármacos anti-TNF (etanercept e infliximab) con resultados favorables y que, además, facilitan la reducción de dosis de corticoides y/o inmunosupresores80. Recientemente se documentó un caso refractario que respondió a tocilizumab, un anticuerpo contra el receptor de la interleucina-681. También es útil la revascularización quirúrgica del vaso afectado.
Panarteritis nudosa cutáneaEs una forma rara de vasculitis de etiología aún desconocida. Es más frecuente en mujeres jóvenes82. Aunque su principal diagnóstico diferencial es la panarteritis nudosa sistémica (PANs), la panarteritis nudosa cutánea (PANc) se encuentra limitada al tejido cutáneo y no progresa a los órganos internos83. Excepcionalmente se ha descrito afectación del sistema musculoesquelético y nervioso periférico82. El curso de la PANc es benigno, crónico y en forma de brotes83. Ha sido relacionada con fármacos e infecciones tales como hepatitis B y C, tuberculosis y estreptocócicas. Es conocida su asociación con enfermedades inflamatorias como la EII (tanto EC84,85 como CU).
La clínica consiste en nódulos dolorosos subcutáneos (fig. 8), livedo reticularis, úlceras cutáneas y necrosis82. Más raramente puede haber púrpura, pápulas, edema o atrofia blanca82. Deja cicatriz postinflamatoria y el cuadro puede ir acompañado de fiebre, artromialgias y parestesias83 (en ocasiones se han documentado parálisis y artritis82). Las lesiones tienen predilección por las extremidades inferiores82.

Histopatológicamente se observa arteritis necrotizante en la dermis profunda y el panículo82. Puede existir oclusión vascular acompañante82, leucocitoclasia y edema. La tinción de van Gieson demuestra destrucción de la lámina elástica interna82. Es obligatorio descartar la PANs debido a su grave pronóstico.
El tratamiento se basa en reposo y antiinflamatorios no esteroideos82. Puede añadirse dapsona82 o colchicina. Deben usarse corticoides sistémicos cuando existe clínica neurológica, muscular o se trata de casos muy recurrentes o refractarios82. En la literatura se menciona la asociación de inmunosupresores (azatioprina, metrotexato, ciclosporina o ciclofosfamida) a la terapia corticoidea sistémica82 y se han publicado casos refractarios tratados con éxito con IGIV. Los pacientes con altos títulos de ASLO se han visto beneficiados de la profilaxis con penicilina82.
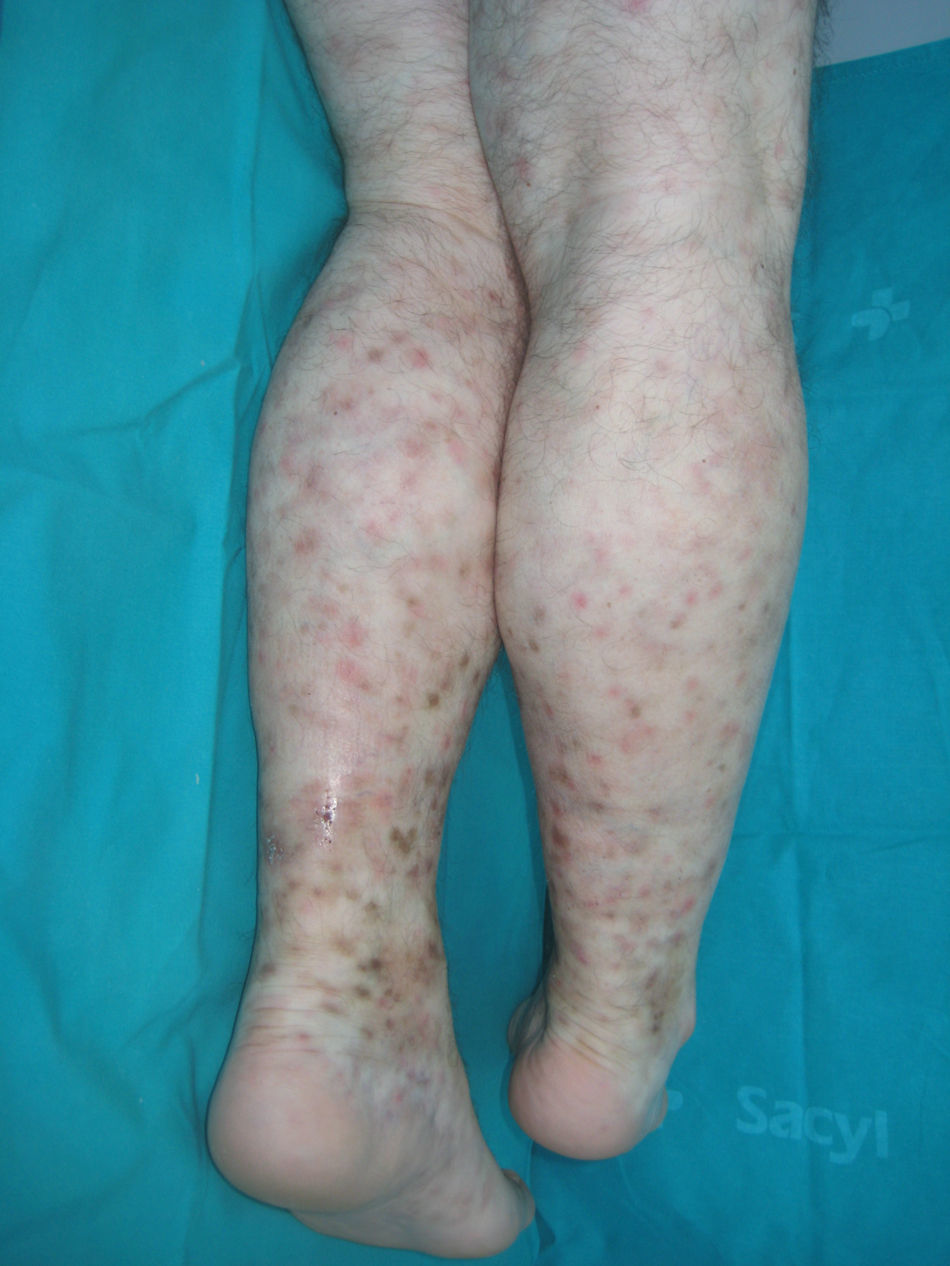
Vasculitis leucocitoclásticaLa vasculitis leucocitoclástica (VL) es la vasculitis más frecuente y afecta predominantemente a vénulas poscapilares86. Tiene mayor prevalencia en mujeres (3:1) jóvenes. Es una reacción de hipersensibilidad antigénica con presencia de inmunocomplejos circulantes86. Son posibles desencadenantes: infecciones, procesos malignos, inflamatorios, autoinmunes y fármacos (infliximab87)86. Se han descrito casos de VL en pacientes con EC88 y CU86. La aparición de VL previa a la CU no es infrecuente86.
La presentación típica es la púrpura palpable en extremidades inferiores (fig. 9)86. Aunque puede tener un inicio macular, lo más común es la presencia de pápulas, placas, nódulos, vesículas, ampollas o pústulas. También son frecuentes lesiones de urticaria, edema o livedo reticularis. El proceso puede desarrollar necrosis, ulceración e hiperpigmentación secundaria. En ocasiones se acompaña de fiebre, astenia, anorexia y artromialgias86. Los síntomas más comunes son dolor, prurito y sensación de quemazón. Existen casos recidivantes que pueden indicar la existencia de una enfermedad subyacente, como crioglobulinemia o infección por hepatitis C.

En la anatomía patológica se aprecian cambios vasculíticos y células polimorfonucleares con fragmentación nuclear (leucocitoclasia o cariorrexis) (fig. 10). Es característica la presencia de tumefacción endotelial y necrosis fibrinoide de las paredes vasculares. En la dermis superficial pueden demostrarse depósitos perivasculares de IgM o C3. La evolución es benigna y el pronóstico es excelente. Generalmente es un trastorno autolimitado y es suficiente el tratamiento sintomático. En casos más agresivos resulta muy eficaz el tratamiento por vía oral con corticoides sistémicos a dosis de 0,5-1mg/kg/d86. Como tratamientos alternativos destacan la dapsona y la colchicina 86. Algunos casos refractarios han sido tratados con azatioprina con éxito. Otros fármacos que han sido beneficiosos son el metrotexato, la talidomida y la pentoxifilina. Se tratará la causa desencadenante en los casos en que se identifique. Cuando se asocia a EII, puede administrase sulfasalazina y colquicina86.
Dermatosis ampollaresDermatosis ampollar IgA lineal
La dermatosis ampollar IgA lineal (DAAL) es una erupción vesiculoampollosa subepidérmica producida por depósitos lineales de IgA a lo largo de la membrana basal (MB). Esta entidad está asociada a infecciones, fármacos, enteropatía sensible al gluten, enfermedades autoinmunes y procesos malignos89. A pesar de ser una dermatosis muy rara, resulta muy llamativa su asociación con la CU (hasta en el 7,1% de los casos en una serie de 70 pacientes con DAAL90). Se han descrito casos aislados en pacientes con EC.
El cuadro clínico es variable. Es característica la disposición de las lesiones «en corona de joyas». Puede presentarse con ampollas tensas con una distribución herpetiforme sobre piel eritematosa o normal en tronco y extremidades89. En ocasiones aparece como una variante del penfigoide cicatricial, afectando la mucosa oral, nasal y esofágica. Existe una forma ocular.
El cuadro histológico muestra un despegamiento subepidérmico con abundantes neutrófilos. En lesiones tempranas, los neutrófilos se alinean en la MB y pueden formar microabscesos en las papilas dérmicas. En ocasiones se aprecian eosinófilos89. El estudio de IF directa revela los depósitos lineales de IgA sobre la MB (membrana lúcida o sublámina densa, según el subtipo). La IF indirecta detecta autoanticuerpos circulantes anti-MB de tipo IgA. Debido a que comparten características clínicas e histológicas comunes, debe descartarse siempre una dermatitis herpetiforme. El tratamiento de elección es la dapsona89 o la sulfapiridina. Aunque existen excepciones90, el tratamiento efectivo de la CU controla generalmente la DAAL90.
Epidermólisis ampollar adquiridaEs una enfermedad crónica autoinmune en la que se forman ampollas tras la lesión del colágeno de tipo VII de la unión dermoepidérmica91. Es adquirida y muy rara. Aunque la epidermólisis ampollar adquirida (EAA) puede asociarse a diversas enfermedades sistémicas, la EC es la más frecuente. Es una enfermedad mecanoampollar: se forman ampollas tras traumatismos. Por este motivo, se ven afectadas las zonas acras y con tendencia a traumatismos, como rodillas, codos y dorso de manos. También puede lesionar las mucosas y el cuero cabelludo hasta provocar una alopecia cicatricial. Las ampollas no son inflamatorias, pueden tener un aspecto hemorrágico y es común que curen con cicatrización, milia e hiper o hipopigmentación postinflamatoria. Son frecuentes las secuelas: sindactilia, distrofia, pérdida de las uñas e impotencia funcional grave. En la microscopia óptica se aprecia un despegamiento subepidérmico con un escaso o nulo infiltrado inflamatorio dérmico de tipo mixto. La IF directa muestra depósitos de IgG lineales en la MB91 y pueden observarse con menor frecuencia depósitos lineales de C3, IgA o IgM. El estudio por IF indirecta detecta autoanticuerpos circulantes anti-MB de tipo IgG y, en ocasiones, IgA91. Por último, mediante estudios de inmunoaglutinación de muestras dérmicas se demuestra que los autoanticuerpos circulantes de estos pacientes se unen a proteínas de 290 kDa y 145 kDa, correspondientes con el colágeno de tipo VII91.
El diagnóstico diferencial de la EAA incluye especialmente la epidermólisis ampollar distrófica, el penfigoide ampollar y el penfigoide cicatricial91. Aunque no se trata de una patología mortal, la calidad de vida de estos pacientes disminuye de forma dramática. No existe ningún tratamiento definitivo y resulta insatisfactorio en la mayoría de la ocasiones91. Se han utilizado dosis altas de corticoides sistémicos y diferentes fármacos inmunosupresores91. Han sido publicados casos que han respondido relativamente a colchicina, dapsona, infliximab e IGIV91,92. Recientemente varios trabajos demuestran la efectividad de rituximab, anticuerpo monoclonal anti-CD20, frente a esta entidad92.
Asociaciones infrecuentesEl «eritema elevatum diutinum» ha sido relacionado más frecuentemente con EC que con CU.
La relación entre liquen plano y EII es controvertida. Aun así, se han publicado casos en los que se sugiere tal asociación.
Liquen nítidus y EC es extremadamente rara.
Se ha descrito excepcionalmente la coexistencia de dermatitis herpetiforme y CU.
La fiebre mediterránea familiar se describió junto a EC en una ocasión.
Ha sido publicado algún caso de EC acompañado de vitíligo.
Acropaquía digital: signo que se correlaciona con la duración, extensión y gravedad de la EII.
- 1.
La enfermedad inflamatoria intestinal (EII) puede presentar afectación extraintestinal en un 20-40% de los casos (tabla 1).
- 2.
Las manifestaciones dermatológicas en pacientes con EII son frecuentes y ocurren hasta en el 10%. Pueden preceder a la aparición de la EII.
- 3.
El eritema nudoso es la única dermatosis relacionada con la actividad de la EII y la más frecuente (afecta hasta un 20%). Es una paniculitis septal sin vasculitis en la que se observan nódulos subcutáneos eritematosos, calientes y dolorosos en extremidades inferiores.
- 4.
El pioderma gangrenoso (PG) es una entidad neutrofílica ulcerosa y recurrente que se asocia a EII hasta en un 12% de los casos.
- 5.
La EII es la enfermedad subyacente más frecuente en el síndrome de Sweet (12%) y todos los pacientes descritos tenían afectación colónica.
- 6.
Es importante conocer las diferentes entidades cutáneas específicas de la EII: piodermatitis-pioestomatitis vegetante (más frecuente en la colitis ulcerosa), granulomatosis orofacial (GOF), enfermedad de Crohn (EC) perianal y EC metastásica.
- 7.
Se ha descrito la asociación de EC y carcinoma: el más frecuente es el adenocarcinoma (59%), seguido por el carcinoma escamoso (31%).
- 8.
La panarteritis nudosa cutánea tiene un curso benigno, crónico y en forma de brotes. A diferencia de la forma sistémica, sólo afecta la piel.
- 9.
Se asocian a EII dos dermatosis ampollares: dermatosis ampollar IgA lineal (DAAL) y epidermólisis ampollar adquirida (EAA). Hasta un 7,1% de pacientes con DAAL presentan colitis ulcerosa. Por su parte, la EC es la entidad más frecuentemente relacionada a la EAA.
- 10.
Recientemente se han publicado casos de pacientes con EII acompañado de alguna estas entidades que, siendo resistentes a tratamientos habituales, respondieron a tratamiento con fármacos biológicos de forma satisfactoria.
Los autores declaran no tener ningún conflicto de intereses.