





Las enfermedades neurodegenerativas constituyen, junto con las enfermedades circulatorias y tumores, la causa de muerte más importante en la población española. En el presente trabajo se aborda el papel que desempeñan los neurotransmisores en la génesis de este tipo de enfermedades, las acciones farmacológicas ante los procesos de muerte celular y la importancia del diagnóstico precoz en el tratamiento.
Dentro de las enfermedades neurodegenerativas destacan los accidentes vasculares, la enfermedad de Alzheimer (EA), la enfermedad de Parkinson (EP) y la esclerosis lateral amiotrófica (ELA). Este tipo de enfermedades se caracterizan por una disminución en el número de células en determinadas poblaciones neuronales. Por ejemplo, en la EA se ha observado una depleción en neuronas colinérgicas del hipocampo, amígdala y corteza, en la EP son las neuronas dopaminérgicas de la sustancia nigra y ganglios basales las afectadas, en enfermedades como la de Huntington son las neuronas de los ganglios basales y del tálamo y, por último, en la ELA se han descrito disminuciones en la población de motoneuronas.
Esta pérdida neuronal se refleja en la aparición de disfuncionalidades tales como alteraciones en los procesos de memoria y lenguaje en la EA, modificación en el control y la coordinación del movimiento en la EP, disminución en las capacidades intelectuales y aparición de movimientos irregulares e involuntarios de las extremidades o de los músculos de la cara en la enfermedad de Huntington y la progresiva paralización de los músculos que intervienen en la movilidad, el habla, la deglución y la respiración en la ELA.
La farmacología, en la búsqueda de nuevos principios activos que interfieran con los procesos neurodegenerativos, mantiene abiertas dos grandes líneas de investigación. La primera intenta prevenir, retardar o paliar la aparición de la sintomatología propia de la alteración en los niveles de neurotransmisores, presentando como objetivo principal su mantenimiento. La segunda centra sus estudios en los procesos de muerte neuronal y en la búsqueda de fármacos que modulen las rutas moleculares y celulares implicadas en estos procesos.
Niveles adecuados de neurotransmisores
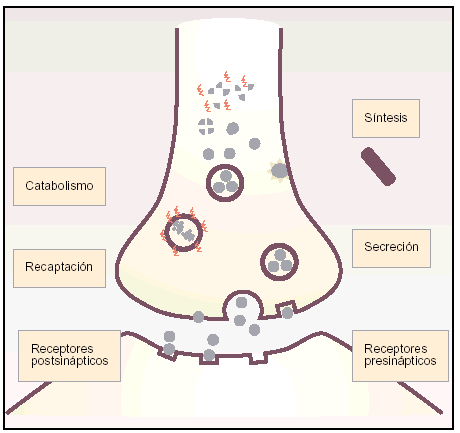
Los neurotransmisores son compuestos químicos que se liberan en la superficie presináptica y se ligan a sus correspondientes receptores de la superficie posináptica produciendo, en algunos casos, un cambio en el potencial de acción posináptico. Se han descrito en determinadas enfermedades neurodegenerativas alteraciones en los niveles de algunos neurotransmisores. Así, se han observado disminuciones de acetilcolina en la EA y de dopamina en la EP, mientras que en la ELA y el ictus se han descrito incrementos en la concentración de glutamato.
Antes de la década de los ochenta se utilizaron psicoestimulantes y vasodilatadores cerebrales orientados al tratamiento de una enfermedad sindromática llamada deterioro mental senil, basada en la insuficiencia cerebral o deterioro cerebral relacionado con la edad. Uno de esos medicamentos fue la dihidroergotoxina, aprobada por la FDA para ese fin. El mejor conocimiento de la fisiopatología, en especial en los últimos 10 años, ha dado las bases a la farmacología para el desarrollo de nuevos fármacos que puedan modular los procesos que regulan la concentración de los neurotransmisores como son: síntesis, almacenamiento, liberación, catabolismo y recaptación en la sinapsis.

Dianas farmacológicas en el mantenimiento de los niveles de neurotransmisores.
Acetilcolina
La acetilcolina es el neurotransmisor específico de las sinapsis, tanto del sistema nervioso somático como en las sinapsis ganglionares del sistema nervioso autónomo y en los órganos diana de la división parasimpática. La síntesis de acetilcolina tiene lugar en el botón terminal y es metabolizada por la acetilcolinesterasa en el espacio sináptico. Ésta participa en la regulación de los niveles de vigilancia y en el funcionamiento de grandes áreas de asociación. Variaciones de sus niveles conllevan alteraciones en la conducta, con síndromes característicos como la pérdida de memoria y atención, el habla confusa y la ataxia, la confusión y la desorientación. Variaciones en las concentraciones de acetilcolina, bien aumentos como disminuciones, han sido descritas en enfermedades como las de Parkinson y Alzheimer, respectivamente.
La acetilcolina ve aumentada su acción cuando desciende el nivel del neurotransmisor dopamina. El bloqueo de la acción de la acetilcolina resulta eficaz en el control del temblor y la rigidez descritos en la enfermedad de Parkinson. Los fármacos anticolinérgicos como el trihexifenidilo, el mesilato de benztropina y la prociclidina son utilizados en esta patología.
Una disminución en los niveles de acetilcolina se ha descrito en pacientes con la EA, por lo que gran parte de los estudios farmacológicos en el tratamiento de esta enfermedad se han dirigido hacia fármacos que incrementen los niveles de dicho neurotransmisor. De forma más concreta, esta línea de investigación presenta tres objetivos:
Fármacos que potencian la síntesis de acetilcolina como lecitina y la CDP colina.
Fármacos agonistas de los receptores nicotínicos como el betanecol, la oxotremonina, la xanomelina y la propia nicotina, entre otros.
Fármacos inhibidores del catabolismo de la acetilcolina, inhibidores de la acetilcolinesterasa como la tacrina, el donepezilo, la rivastigmina y la galantamina.
De todos ellos, los más utilizados son los fármacos inhibidores de la acetilcolinesterasa (o anticolinesterásicos) que son capaces de inhibir el catabolismo del neurotransmisor y aumentar la permanencia de éste en la sinapsis. El primer fármaco utilizado fue la tacrina, un inhibidor alostérico reversible, que ha dejado de ser empleado en clínica debido a su efecto hepatotóxico. En la actualidad disponemos de fármacos más seguros, como el donepezilo, derivado de las piperidinas, que es inhibidor reversible no competitivo mixto y la rivastigmina, un inhibidor pseudoirreversible no competitivo. De reciente introducción es la galantamina, un inhibidor reversible competitivo que a su vez es un modulador alostérico del receptor nicotínico, lo que hace que aumente la liberación de acetilcolina en la sinapsis. La galantamina, cuya eficacia clínica ha sido probada, mantiene la función cognitiva y el funcionamiento diario a largo plazo, postergando la aparición de las alteraciones de la conducta. En general, este grupo de fármacos mejora de forma leve o moderada los trastornos cognitivos y conductuales en pacientes con EA aunque ninguno de ellos detiene la evolución de la enfermedad.
Una segunda la línea de investigación está dirigida a buscar fármacos que activen los receptores colinérgicos de tipo muscarínico y nicotínico, tanto a nivel pre como posináptico. Se han estudiado varios agonistas que actúan sobre los receptores muscarínicos como la pilocarpina, la arecolina y la oxotremolina entre otros, pero los resultados no han sido favorables. Hoy día se investigan otros como la xalomelina y la milamelina, pero aún están en fase II de investigación. Los estudios sobre los receptores nicotínicos son contradictorios y no se puede afirmar, de momento, que los parches de nicotina o la nicotina per se puedan producir beneficios en la terapéutica específica en la EA. En algunos estudios de prevalencia de demencia tipo Alzheimer se encontró que la población de fumadores podría tener un factor protector, pero este dato ha sido corregido, entre otros aspectos por el mayor riesgo de poder presentar una demencia vascular.
Dopamina
Al presentar una localización encefálica más elevada que la noradrenalina, la dopamina es el neurotransmisor catecolaminérgico más importante y, por tanto, con mayor repercusión comportamental. Disminuciones en los niveles de dopamina se han descrito en cerebros post mortem, provenientes de pacientes con enfermedad de Parkinson. La farmacología mantiene abiertos cuatro frentes de investigación con un único objetivo, la recuperación de los niveles de dopamina. Así, investiga sobre fármacos:
La levodopa es capaz de atravesar la barrera hematoencefálica, siendo el fármaco más eficaz en el tratamiento de la enfermedad de Parkinson
Precursores de la síntesis de dopamina, como la levodopa.
Potenciadores de su liberación en los botones sinápticos, como la amantadina.
Agonistas de receptores dopaminérgicos, como la bromocriptina, la apomorfina, la lisurida, el soligen y la pergolida.
Inhibidores de las enzimas encargadas de su catabolismo, como la monoamino oxidasa B (MAO B), destacando la selegilina, o la enzima catecol-O-metiltransferasa (COMT); la entacapona y la tolcapona son dos fármacos de este grupo.
Debido a que la dopamina no atraviesa la barrera hematoencefálica, no se utiliza como fármaco. Sin embargo, son varias las aproximaciones que se han realizado con el fin de aumentar directamente sus niveles, fundamentalmente mediante trasplantes en las áreas afectadas de células capaces de secretar dopamina como son neuronas dopaminérgicas, células cromafines de la glándula suprarrenal y células del cuerpo carotídeo. Recientemente, y en modelos animales, se están realizando trasplantes de células indiferenciadas logrando su crecimiento y diferenciación completa hasta neuronas dopaminérgicas.
El uso de precursores de dopamina es ampliamente utilizado y entre ellos destaca la levodopa. La levodopa es capaz de atravesar la barrera hematoencefálica, siendo el fármaco más eficaz en el tratamiento de la enfermedad de Parkinson. La levodopa se absorbe bien en el intestino delgado, aunque es inactivada en una gran proporción, próxima al 95% en algunos casos, por la MAO periférica, dando lugar a la aparición de efectos secundarios conocidos. Con el fin de evitar esta transformación periférica se suele coadministrar junto a fármacos inhibidores de la descarboxilasa, como la carbidopa o la benseracida, incapaces de atravesar la barrera hematoencefálica, y de esta manera no sólo se evitan los efectos secundarios sino que se reduce unas 10 veces las dosis de levodopa necesarias.
Una segunda aproximación farmacológica centra sus objetivos en aumentar la liberación del neurotransmisor. Éste es el caso del clorhidrato de amantadina, originariamente utilizado como fármaco antiviral para la profilaxis y el tratamiento de infecciones producidas por la gripe de tipo A, aumenta la liberación y bloquea la recaptación de la dopamina. La amantadina se ha utilizado en estadios iniciales de la enfermedad de Parkinson, tanto en monoterapia como asociada a levodopa/carbidopa de forma intermitente.
Los fármacos agonistas de receptores dopaminérgicos constituyen la tercera aproximación. La bromocriptina y la pergolida mimetizan la función de la dopamina y son utilizados tanto en las etapas iniciales de la enfermedad como para prolongar la respuesta a la levodopa en los enfermos que sufren una aparición o desaparición repentina de los síntomas. Pueden administrarse de forma conjunta con levodopa, ya que son menos eficaces que ésta en el control de la rigidez y la bradicinesia. De reciente introducción es el sogilen, un potente agonista de los receptores de dopamina D2 cuya acción es más prolongada que la de cualquier otro tratamiento antiparkinsoniano, proporcionando una eficacia en el control de los síntomas de la EP y de las fluctuaciones motoras durante 24 horas mediante una única dosis diaria.

Por último, citaremos los fármacos inhibidores del catabolismo de la dopamina, en concreto de dos enzimas la MAO B y la COMT. La selegilina, también conocida como deprenil, es un IMAO. Su administración retrasa, en algunos casos, la implantación del tratamiento con levodopa. Ensayos clínicos han puesto de manifiesto que la combinación de selegilina con levodopa, a largo plazo, resulta más eficaz para controlar los síntomas y aumentar la supervivencia que la administración de levodopa sola. Los fármacos inhibidores de la COMT de segunda generación presentan la característica de ser más selectivos y activos por vía oral, destacando los derivados del dinitrofenol, como la entacapona y la tolcapona. En la actualidad, se están desarrollando varios ensayos clínicos que evalúan la combinación de un ICOMT como la entacapona con la levodopa.
Glutamato
El glutamato es el neurotransmisor excitatorio por excelencia. Media su efecto activando distintos receptores: ionotrópicos, que forman canales iónicos de localización principalmente postsináptica como el receptor de NMDA (N-metil-D-aspartato), vinculado al flujo de cationes Na+, K+, Ca2+, el receptor de AMPA y el receptor para kainato, asociado a los iones Na+ y K+, y otros, presinápticos, conocidos como metabotrópicos, al mediar su efecto mediante la activación de proteínas G.
Aunque el glutamato desempeña importantes funciones fisiológicas en el SNC, participa en el 70% de las sinapsis excitatorias, la activación excesiva de sus receptores, bajo ciertas condiciones, es neurotóxica, habiéndose relacionado con procesos neurodegenerativos tanto agudos como crónicos. En la neurotoxicidad aguda (por ejemplo, isquemia cerebral) el déficit energético celular conlleva a una disminución en la recaptación de glutamato del espacio sináptico, al consecuente aumento en su concentración y a un incremento de la sensibilidad de las neuronas al glutamato. La sobreactivación de los receptores en esta situación tiene como resultado una alteración en los iones Na+ y Cl que genera la entrada masiva de agua, induciendo la lisis osmótica de la neurona. Por otro lado, la neurotoxicidad crónica está mediada por mecanismos dependientes de la entrada de Ca2+ y está implicada en enfermedades como la esclerosis lateral amiotrófica, la demencia asociada al sida, la enfermedad de Parkinson, la enfermedad de Huntington y la enfermedad de Alzheimer.
La «farmacología del glutamato» ha centrado sus estudios en fármacos que, o bien disminuyen los niveles sinápticos del neurotransmisor (riluzol, lubeluzol), o antagonizan los receptores ionotrópicos (selfotel, eliptorodil, dizocilpina y gavestigel, entre otros).
Aunque el mecanismo exacto de acción del riluzol se desconoce, éste puede mediar su efecto por la inhibición de los canales de sodio dependiente de voltaje y, de esta manera, impedir la entrada masiva de Ca2+ en la neurona presináptica, disminuyendo la secreción de neurotransmisor. El riluzol es aceptado para el tratamiento de la ELA y se encuentran abiertos varios ensayos clínicos en pacientes de EA, EP, EH e ictus.
Entre los fármacos antagonistas del receptor de NMDA destacan el selfotel (no competitivo reversible); el eliptorodil (un bloqueador del lugar de unión de las poliaminas); la dizocilpina (inhibidor no competitivo), el gavestigel (que se une al lugar de unión de la glicina) y la memantina (antagonista no competitivo). La utilización en clínica de este grupo de fármacos es complicada. Ensayos clínicos sobre neuroprotección, en pacientes que habían sufrido isquemia cerebral, tuvieron que ser detenidos, bien por un resultado desfavorable en la relación riesgo/beneficio como fue el caso de selfotel o cerestat, o por una evolución funcional negativa, como la encontrada con el eliprodil y el GV150526. La memantina presenta una actividad neuroprotectora y es el primero de su grupo utilizado en el tratamiento de la EA. La memantina puede mejorar los síntomas de la demencia (mejoría funcional y reducción de la dependencia) en pacientes con enfermedad moderada a grave.
Los procesos de muerte celular como diana farmacológica
Los procesos de muerte celular se engloban dentro de dos grandes bloques: necrosis y apoptosis. La necrosis se ha relacionado con estados agudos donde la célula pierde, de una forma brusca, la integridad de sus organelas, disipando la capacidad de síntesis de energía y el control de la homeostasis celular. Estos procesos se han relacionado con patologías como el trauma o la isquemia. Sin embargo, durante los procesos apoptóticos la célula mantiene la integridad de sus organelas hasta estadios bien tardíos, lo que le permite conservar el estado energético, la homeostasis celular y, en algunos casos, mantener la maquinaria necesaria para sintetizar proteínas implicadas en estas rutas, por lo que también se le ha denominado muerte celular programada. Células con características apoptóticas se han observado en áreas afectadas en neuropatías como EA, ELA, EP, EH, entre otras.
En los procesos apoptóticos se pueden distinguir tres fases: activación, propagación y ejecución, que, en ocasiones, son precedidas por un período de latencia, de extensión variable. La fase de activación suele estar mediada por señales extracelulares, como la unión de un ligando a su receptor; sirva de ejemplo el factor de necrosis tumoral alfa (TNF-alfa) o los neurotransmisores excitatorios, antes comentados. La fase de propagación, ya intracelular, está mediada por segundos mensajeros, como el Ca2+, las especies reactivas del oxígeno (ERO) y factores de transcripción como p53 y AP-1, entre otros. Finalmente, en la etapa de ejecución, la célula activa procesos de degradación en los que participan proteasas y ADNasas. Entre las dos últimas etapas se ha situado a la mitocondria como organela clave en el punto de no retorno.
En los siguientes apartados analizaremos las tres fases de los procesos apoptóticos: activación, propagación y ejecución.

El calcio en los procesos neurodegenerativos.
Fase de activación
Calcio
El calcio (Ca2+) es el ion fundamental en los procesos intracelulares de las células excitables incluyendo las neuronas. En el sistema nervioso central, el Ca2+ interviene en multitud de procesos, incluyendo la producción del potencial de acción, la liberación de neurotransmisores, la plasticidad neuronal y probablemente la formación de memoria. Sin embargo, la entrada masiva de Ca2+ o el fallo de los mecanismos que regulan su concentración intracelular desencadena una serie de acontecimientos que conlleva la muerte neuronal. En los últimos veinte años, ha tomado cuerpo una «teoría unificada» que pretende explicar cómo una serie de distintas patologías, desde la isquemia cerebral hasta las enfermedades neurodegenerativas, confluye en último término en la muerte neuronal mediada directa o indirectamente por Ca2+. El aumento de la concentración de calcio intraneuronal puede conducir a la activación de rutas moleculares, entre las que se incluyen las proteincinasas, las fosfolipasas A2, la óxido nítrico sintetasa y las proteasas. En algunos casos, estas rutas inducen la generación de radicales libres, con el consiguiente daño en organelas como la mitocondria.
La homeostasis del calcio es resultado del equilibrio de sistemas que regulan su entrada, como los receptores de glutamato y los canales de calcio dependientes de voltaje (CCDV) y sistemas encargados en su extrusión como las bombas dependientes de ATP y organelas con capacidad de retenerlo en su interior como la mitocondria y el retículo endoplasmático.
Los fármacos inhibidores de los CCDV, como el nimodipino, el nicardipino y la flunaricina, empezaron a ser utilizados en procesos neurodegenerativos tales como la enfermedad de Alzheimer y el ictus, quizá debido a los resultados esperanzadores obtenidos en modelos experimentales. Sin embargo, los resultados obtenidos en los ensayos clínicos no fueron los deseados. Así, el nimodipino, administrado por vía endovenosa en pacientes que han padecido una isquemia cerebral focal, presentó resultados dispares desde una mejoría en evolución funcional frente a placebo, hasta no encontrar diferencias; es más, alguno de estos ensayos tuvo que ser suspendido prematuramente bien por razones de seguridad, como por una peor evolución del grupo activo. En esta misma patología se han detenido ensayos clínicos con nicardipino, isradipino, darodipino y flunaricina
Las neuronas, en su citoplasma, contienen proteínas con capacidad de amortiguar Ca2+ como la calbindina o la parvoalbúmina. En determinadas patologías, como la EA y la ELA, los grupos neuronales que carecen o expresan en cantidades muy pequeñas estas proteínas resultan más sensibles, mientras que aquellos que expresan grandes cantidades parecen estar relativamente respetados. En nuestro grupo de investigación utilizamos vectores adenovirales, con el fin de sobreexpresar la calbindina D28K en cultivos primarios de neuronas de rata. La sobreexpresión de esta proteína ha conferido protección a estos cultivos celulares frente a estímulos como la presencia de péptido amiliode 25-35 o la adición de un fármaco, ampliamente utilizado como modelo inductor de apoptosis, como es la estaurosporina. Sin embargo, ésta no confirió protección a cultivos expuestos a radicaciones gamma o en aquellos donde el soporte trófico era retirado.
Especies reactivas del oxígeno
Aunque el estado de estrés oxidativo no es el causante directo o el factor etiológico responsable de las neuropatologías, existen evidencias de que la presencia de radicales libres produce el daño tisular y degenerativo secundario que acompaña a estas enfermedades. El sistema nervioso central, y en concreto el cerebro, tiene un alto requerimiento metabólico de oxígeno y una alta concentración de ácidos grasos poliinsaturados, lo que le convierte en un lugar propenso a la peroxidación lipídica. La sobreproducción de radicales libres que pueden atacar y dañar irremediablemente el tejido neuronal contribuye de manera directa o sinérgica al proceso neurodegenerativo. Para prevenir los aumentos descontrolados de EROS las células tienen sistemas antioxidantes. Estos sistemas pueden clasificarse en sistemas enzimáticos (superóxido dismutasa, la catalasa y la glutatión peroxidasa), antioxidantes endógenos (trasferrina, la lactoferrina, la ceruloplamina, la albúmina, la bilirrubina, el ácido úrico) o contenidos en la dieta (las vitaminas C y E, el betacaroteno, los flavonoides, el selenio y el cinc) y, por último, antioxidantes farmacológicos (tirilazad, ebselen, N-acetilcisteína, porfirinas), entre otros.
El mantenimiento y la optimización de los mecanismos de defensa antioxidante en el cerebro constituyen una estrategia importante para prevenir o reducir la progresión de la disminución de la neurodegeneración. El potencial de los antioxidantes dietarios --incluidos el betacaroteno, la vitamina C y la vitamina E-- para que protejan contra la pérdida cognitiva y los desórdenes neurodegenerativos ha sido explorado en varios estudios epidemiológicos y clínicos, mostrando una correlación significativa entre los niveles de vitamina E en el suero y la memoria en una población adulta mayor. Específicamente, existen resultados de cómo los niveles de vitamina E se asocian a la pérdida cognitiva relacionada con la edad y la EA. Asimismo, ésta ha resultado eficaz en modelos experimentales, retardando la aparición de las alteraciones motoras de la ELA y neuroprotectora en cultivos neuronales.
Durante algunos procesos de muerte celular se produce la activación de rutas metabólicas que necesitan de la síntesis de novo de determinadas proteínas
Pacientes que han sufrido una isquemia cerebral y han sido tratados con fármacos antioxidantes como Tirilazad y Ebselen presentaron una evolución funcional, aunque no una mejoría significativa. El uso de antioxidantes, como la coenzima Q10 y el tocoferol, así como de suplementos nutricionales, como la creatina y la nicotinamida, además de la restricción nutricional del hierro y el uso de quelantes del hierro, merecen especial atención como estrategias en la prevención de la neurodegeneración de la enfermedad de Huntington.
Factores de transcripción
Durante algunos procesos de muerte celular se produce la activación de rutas metabólicas que necesitan de la síntesis de novo de determinadas proteínas. Una de las formas más importantes de control de la expresión génica es la regulación transcripcional, y dentro de ésta, los factores de transcripción (FT) se encargan de llevarla a cabo de forma preferente. Los FT son proteínas que se unen a secuencias específicas de DNA y que modulan la expresión de los genes. Los FT se encuentran localizados en el citoplasma de forma inactiva y, tras un estímulo determinado, Ca2+, EROS o factores tróficos son activados traslocándose al núcleo. Entre los factores de transcripción destacan el factor nuclear κB (NF-κB), la familia de la proteína activadora-1 (AP-1), que engloba miembros como c-fos, c-jun, y el p53, entre otros.
El enorme progreso en la comprensión de los mecanismos de transcripción ofrece la posibilidad del desarrollo de una nueva generación de fármacos, que deberán ser capaces de modular la síntesis de factores de transcripción, regular su actividad y las interacciones con proteínas activadoras e inhibidoras o su unión a ADN. Debido a la alta especificidad de esta nueva vía terapéutica, parece claro que estos agentes proporcionarán buenas herramientas para la prevención y el tratamiento de una gran variedad de enfermedades en un futuro no muy lejano.
Uno de los más importantes es el factor nuclear κB (NF-κB) que permanece en el citoplasma celular unido a proteínas inhibitorias conocidas como IkBs. Cuando una señal externa, como factores neurotróficos o EROS, afecta a la célula el complejo IkB cinasa (IKK) se activa y fosforila a las proteínas IkB. Entonces, IkB fosforilada es degradada rápidamente, liberando y activando así al NF-kB. Entre los fármacos inhibidores de la familia NF-κB, se encuentran algunos de los fármacos antiinflamatorios más comúnmente usados, como es el salicilato aspirina y algunos esteroides.
Otra diana farmacológica es la proteína p53. Ésta controla la integridad del ADN y la terminación correcta de las diferentes fase del ciclo, deteniendo el crecimiento celular cuando existe daño en el ADN o los sistemas de control se desregulan. Se ha encontrado activada en pacientes de la EP, EA, y tras la exposición a radiaciones gamma. Fármacos inhibidores de p53, como la pifithrin-alfa y el Z-1-117, han sido eficaces en modelos experimentales frente a diversos estímulos neurotóxicos.
Fase de propagación
La mitocondria, que durante muchos años se consideró sólo como centro generador de energía, ocupa hoy un lugar privilegiado en los procesos de muerte celular, papel que ha sido objeto de revisión por nuestro grupo en esta revista.
Alteraciones mitocondriales como la pérdida del potencial eléctrico mitocondrial, alteraciones en los complejos de la cadena transportadora de electrones, han sido descritas en células de individuos que padecen la corea de Huntington o la enfermedad de Alzheimer. El campo de la farmacología mitocondrial se ha centrado en los últimos años en el estudio de los procesos que rodean a la formación y apertura del poro de permeabilidad transitoria mitocondrial (PPTM). El bloqueo de la formación del PPTM resulta en la protección de cultivos celulares frente a estímulos apoptóticos que pueden ser mediados por el Ca2+ y las EROS. Fármacos bloqueadores de la apertura del PPTM, como la ciclosporina A y el ácido bongkrekico, presentan efectos neuroprotectores en modelos neurotóxicos de la corea de Huntington o en las zonas del hipocampo afectadas tras procesos isquémicos o tras la exposición a NMDA.
Fase de ejecución
Durante la fase de ejecución se lleva a cabo la degradación de proteínas, que esta mediada fundamentalmente por la acción de proteasas como los miembros de las familias de cisteína (caspasas y calpaínas) y serina proteasas. Estas enzimas hidrolizan sustratos selectivos como elementos del citoesqueleto (actina, fodrina, proteína tau y catenina), enzimas encargadas de reparar (PARP) o degradar (ADNasa) el ADN celular, factores de transcripción (retinoblastoma, MDM2), y proteínas reguladoras cinasas y fosfatasas (proteína cinasa C, fosfatasas 2A, cinasas de adhesión focal, Akt, raf1).
En modelos experimentales, la activación de las caspasas parece estar implicada en algunas enfermedades neurodegenerativas como la enfermedad de Alzheimer, la esclerosis lateral amiotrófica, la enfermedad de Parkinson y la corea de Huntington. Hoy día, disponemos de fármacos inhibidores de caspasas, tanto de origen viral (crmA, p35 y el péptido inhibidor de apoptosis), como de origen sintético (Z-VAD-FMK específico para las caspasas 1, 3, 4 y 7; Z-DEVD-FMK, inhibidor de la caspasa-3). La utilización de estos fármacos protege a los cultivos celulares de la disminución de la viabilidad inducida por neurotoxinas, y se está comenzando a utilizar, con cierto éxito, en pacientes con enfermedad de Parkinson. Así, el péptido inhibidor Ac-YVAD-CMK favorece la viabilidad de los implantes de neuronas dopaminérgicas y mejora sustancialmente la recuperación funcional de ratas parkinsonianas.
Las calpaínas se han descrito estar activadas en procesos neurodegenerativos como en las neuritas distróficas de placas seniles de pacientes de EA, y se postula que podría ser causa de la pérdida sináptica. Fármacos inhibidores de éstas, como el MDL28170, promueven la recuperación de las respuestas sinápticas en rodajas de hipocampo sometidas a hipoxia y se muestran eficaces frente a los procesos de muerte neuronal inducidos por el péptido betaA(25-35); o los compuestos ALLnL y ALLnM que protegen líneas celulares frente a estímulos como el NMDA y la glucoproteína del virus del sida gp120 y los derivados del alfa-mercaptoacrilato, como el PD150606, que confieren protección a cultivos neuronales cerebro-corticales frente al daño inducido por estímulos hipóxicos/hipoglucémicos.
Otras dianas
Estrategia antiinflamatoria
La progresión de algunas enfermedades neurodegenerativas viene acompañada de procesos de inflamación en astrocitos y microglía. Así, la EA presenta procesos de inflamación asociados a las placas neuríticas y a la degeneración neurofibrilar. Estudios epidemiológicos asocian a los AINE con retrasos en la aparición, o con una progresión más lenta de la EA. Este efecto neuroprotector puede ser explicado por la inhibición de la ciclooxigenasa (COX), y en concreto de la COX-2, cuya expresión se encuentra elevada en áreas relacionadas con la memoria y que se correlaciona con la deposición de la proteína betaamiloide.
Otro fármaco que se encuentra en las primeras fases de investigación es la propentofilina, que interfiere con la neuroinflamación relativa a la activación de las células gliales en la EA y en la demencia vascular.
Homeostasis del colesterol
El tratamiento prolongado con hipolipemiantes, como las estatinas, se ha asociado a una menor incidencia de EA. Los científicos han observado que los niveles de colesterol influyen en la formación de las placas amiloides de la enfermedad de Alzheimer y se han puesto en marcha ensayos para valorar la acción neuroprotectora de las estatinas. Los estudios, coordinados por el Prof. Tobias Hartmann, de la Universidad de Heidelberg (Alemania), señalan que dos medicamentos utilizados para reducir el colesterol, la simvastatina y la lovastatina, son capaces de disminuir notablemente los niveles del péptido betaamiloide. En ratones de laboratorio, la administración de dosis elevadas de simvastatina es capaz de disminuir considerablemente los niveles de los péptidos betaamiloides en el líquido cefalorraquídeo y en los tejidos cerebrales. Los investigadores argumentan que la reducción del colesterol es la causa de la disminución en los niveles de péptidos betaamiloides, y consideran que las estatinas pueden regular los niveles del péptido betaamiloide. Sin embargo, los resultados de otro estudio que utiliza a 2.581 personas, a partir de 800 familias registradas en 15 centros, en el período de 1996 a 2000, no mostraron diferencia en la relación entre el uso de estatinas y el riesgo de aparición de la EA.
Factores de crecimiento y hormonas
La falta o escasez de determinados factores de crecimiento (FC) puede ser causa de procesos neurodegenerativos. El factor de crecimiento nervioso (NGF), la neurotrofina 3 (NT-3), o el factor neurotrófico derivado de cerebro (BDNF), son miembros de la familia genética de las neurotrofinas que inducen supervivencia, diferenciación, mantenimiento y reparación de poblaciones neuronales específicas. Posiblemente la falta de modulación, de los complejos ciclinas-cdk, por parte de determinados FC sea uno de los motivos que conducen a la apoptosis. Además, la ausencia de inducción de los genes responsables de la síntesis de determinadas enzimas antioxidantes, o de determinadas proteínas amortiguadoras de Ca2+, probablemente también influya en el desarrollo de la muerte neuronal. Así, los niveles de BDNF están significativamente reducidos en el hipocampo y la corteza parietal de pacientes con EA. En este sentido, el suministro de determinados FC, como el NGF, la NT-3, o el BDNF, podría constituir una terapia protectora en muchas enfermedades neurodegenerativas.
El factor de crecimiento similar a la insulina (IGF-I) es efectivo en modelos animales de ELA, al igual que el CNTF, el BDNF y la NT-3. Por otra parte, el NGF ha exhibido un efecto protector sobre neuronas colinérgicas en cultivo, que son las mismas que se comprometen tempranamente en la EA, y en la EP experimental se ha mostrado el beneficio del FGF, del TGF-beta y del GDNF.

Diversas pruebas terapéuticas han sido completadas. En pacientes con la ELA se han ensayado el CNTF, el IGF-I y el BDNF sin que hayan logrado modificar el curso natural de la enfermedad. Sin embargo, cabe señalar que en todos esos intentos la vía de administración utilizada ha sido la subcutánea que, aparentemente, no es la más apropiada. Muy posiblemente, la modificación de la vía de administración de estos factores conduzca a resultados más alentadores, como apuntan estudios experimentales que utilizan trasplantes de células modificadas genéticamente capaces de sintetizar FT o vectores virales.
En la actualidad se están desarrollando fármacos capaces de inducir la síntesis de estos FC. El SR577746A aumenta la secreción de neurotrofinas y ha mejorado las alteraciones de la memoria, por lo que podría ser éste un candidato para la prevención de EA. El xaliproden, fármaco experimental para la ALS, ha mostrado tener un mayor período de respiración independiente y de supervivencia, al aumentar la producción de factores neurotróficos. Por último, una estimulación moderada de los receptores glutamato puede resultar también en un aumento de la síntesis de FC. La ensaculina, una benzopirona que activa los receptores N-metil-D-aspartato, ha mostrado alguna eficacia en animales de experimentación con respecto a parámetros de memoria, así como una tolerancia aceptable en varones humanos.
La terapia hormonal sustitutiva es también utilizada en pacientes con enfermedades neurodegenerativas. El 17-betaestradiol, además de ejercer sus características acciones hormonales relacionadas con el desarrollo y vida sexual de la mujer, ejerce efectos diversos celulares en el sistema nervioso central, tanto de la mujer como del varón. Los estrógenos poseen acciones de carácter neurotrófico y neuroprotector. Estudios retrospectivos han sugerido que el tratamiento estrogénico en mujeres menopáusicas parece reducir, del 40-50%, el riesgo de padecer la EA. No obstante, se necesitan ensayos clínicos aleatorizados para demostrar la eficacia de la terapia sustitutiva estrogénica en la prevención de la enfermedad. Estudios recientes realizados por investigadores estadounidenses mostraron que una terapia sustitutiva basada en estrógenos administrada durante un año, no redujo la progresión de la enfermedad, ni mejoró la función cognitiva global de 97 pacientes con el mal de Alzheimer. Tratamientos con otras hormonas esteroideas, como el colesterol y la corticosterona, no han conseguido estos resultados.
Otras aproximaciones
El esclarecimiento de las rutas de síntesis del péptido betaamiloide, componente mayoritario de las placas y depósitos neurofibrilares, ha permitido grandes avances en la terapéutica de la EA. El bloqueo del paso de la proteína precursora del betaamiloide (APP) a betaamiloide mediante el uso de fármacos inhibidores de las secretasas, proteasas encargadas de metabolizar la APP, constituye una de las grandes apuestas.
Por último, se han realizado aproximaciones para la construcción de vacunas contra las enfermedades neurodegenerativas. Así, en la EA se ha desarrollado una vacuna experimental, la AN-1792, utilizando placas amiloides como agente invasor del cerebro. Aunque, en un principio, presentó resultados muy esperanzadores ya que su administración a un grupo de pacientes indujo la formación y acumulación de anticuerpos dirigidos contra las placas de betaamiloide, el ensayo clínico ha tenido que ser detenido debido a la aparición de repuesta inflamatoria en el SNC.
Conclusiones
El carácter progresivo de las enfermedades neurodegenerativas es actualmente el mayor reto terapéutico.
La farmacología dispone de una amplia gama de dianas celulares sobre las que actuar bien inhibiéndolas o activándolas, y aunque los resultados obtenidos en los laboratorios de investigación son muy prometedores, sólo los ensayos clínicos nos permitirán el poder aceptar o no un fármaco.
Un diagnóstico precoz resulta clave en el tratamiento de la enfermedad, ya que los enfermos pueden beneficiarse de los fármacos que retardan la aparición de las alteraciones.

Bibliografía general
Aisen PS. Evaluation of selective COX-2 inhibitors for the treatment of Alzheimer's disease. J Pain Symptom Manage 2002;23:35-40.
Arlt S, Beisiegel U, Kontush A. Lipid peroxidation in neurodegeneration: new insights into Alzheimer's disease. Curr Opin Lipidol 2002;13:289-94.
Chen ZY, Cao L, Wang LM, Guo C, Ye JL, Chai YF, Yan ZY. Development of neurotrophic molecules for treatment of neurodegeneration. Curr Protein Pept Sci 2001;2(3):261-76.
Giménez-Roldán S. Agonistas dopaminérgicos en el tratamiento de la enfermedad de Parkinson. Jano 1999;1299:56-9.
González Moneo MJ, Garrido Barral, A. Novedades terapéuticas en el tratamiento de las demencias. Atención Primaria 2002;08(29):507-12.
Heemskerk J, Tobin AJ, Ravina B. From chemical to drug: neurodegeneration drug screening and the ethics of clinical trials. Nat Neurosci 2002;5(supl.202);1027-9.
Jordán J, Galindo MF, Ceña V, González-García C. Cisteína proteasas y neurodegeneración. Rev Neurol 3 2000;333-40.
Lozano JA. El parkinsonismo y su tratamiento. Offarm 2001;6(20):96-106.
Moosmann B, Behl C. 2002. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs 2002;11:1407-35.
Paredes F, Roca JJ. Influencia de los radicales libres en el envejecimiento celular. Offarm 2002;21(7):96-100.
Quik M, Kulak JM. Nicotine and nicotinic receptors; relevance to Parkinson's disease. Neurotoxicology 2002;23:581-94.
Tornero D, Ceña V, González-García C, Jordán J. Papel del poro de permeabilidad transitoria mitocondrial en los procesos neurodegenerativos. Rev Neurol 2002;35:354-61.
Tornero D, Ceña V, Jordán J. La mitocondria como diana farmacológica en los procesos neurodegenerativos. Offarm 2002;21(11):126-30.