




En los últimos años se ha realizado un gran esfuerzo por desarrollar distintas técnicas moleculares para el diagnóstico de laboratorio de las micosis. En este sentido, la elucidación de la secuencia genómica completa de los principales patógenos fúngicos representa una oportunidad sin precedentes para los micólogos clínicos. Existe, además, una gran expectación sobre la posible aplicación de los rápidos avances en el campo de la genómica y la proteómica al estudio de la base molecular de la patogenicidad fúngica, la búsqueda de nuevos antifúngicos más eficaces y la identificación de antígenos fúngicos.
En este artículo se revisan los principales hitos en la genómica de los hongos. Además, se presentan algunas aplicaciones de la genómica y la posgenómica de interés para el micólogo clínico.
Great effort has been made in recent years to develop molecular techniques for laboratory diagnosis of mycosis. To this end, elucidation of the entire genomic sequence of the main fungal pathogens provides an unprecedented opportunity for clinical mycologists. There is considerable expectation regarding the potential application of the rapid advances in genomics and proteomics to study the molecular basis of fungal pathogenicity, to search for new and more effective antifungal drugs, and to identify fungal antigens.
This article reviews the key aspects of fungal genomics and presents genomic and post-genomic applications of interest for clinical mycologists.
Las técnicas tradicionales de diagnóstico micológico se enfrentan con bastante frecuencia a problemas de falta de especificidad, falta de sensibilidad, dificultades técnicas o de interpretación y lentitud1,2. El intento por solventar en la medida de lo posible dichos problemas ha sido un factor crucial para el desarrollo de las técnicas inmunológicas y moleculares.
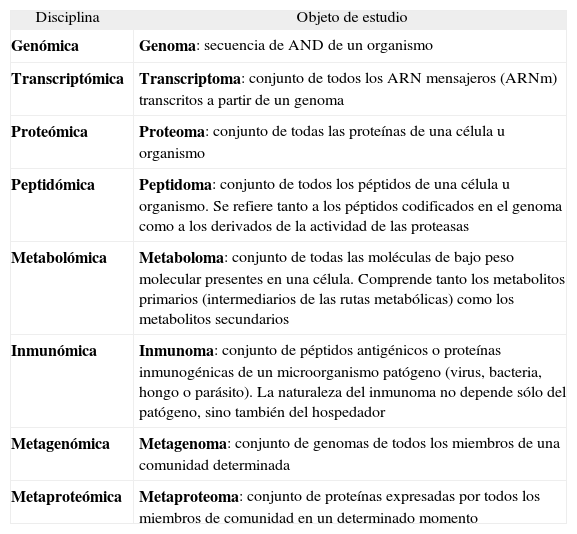
Por otro lado, hemos de situarnos en el contexto de las llamadas "-ómicas", es decir, de las disciplinas científicas que abordan su objeto de estudio de una manera global, como pueden ser la genómica, la proteómica, la metabolómica y otros campos afines. En la tabla 1 se presenta una relación de las principales "-ómicas" y su objeto de estudio.
Principales “-ómicas” y objeto de estudio
| Disciplina | Objeto de estudio |
| Genómica | Genoma: secuencia de AND de un organismo |
| Transcriptómica | Transcriptoma: conjunto de todos los ARN mensajeros (ARNm) transcritos a partir de un genoma |
| Proteómica | Proteoma: conjunto de todas las proteínas de una célula u organismo |
| Peptidómica | Peptidoma: conjunto de todos los péptidos de una célula u organismo. Se refiere tanto a los péptidos codificados en el genoma como a los derivados de la actividad de las proteasas |
| Metabolómica | Metaboloma: conjunto de todas las moléculas de bajo peso molecular presentes en una célula. Comprende tanto los metabolitos primarios (intermediarios de las rutas metabólicas) como los metabolitos secundarios |
| Inmunómica | Inmunoma: conjunto de péptidos antigénicos o proteínas inmunogénicas de un microorganismo patógeno (virus, bacteria, hongo o parásito). La naturaleza del inmunoma no depende sólo del patógeno, sino también del hospedador |
| Metagenómica | Metagenoma: conjunto de genomas de todos los miembros de una comunidad determinada |
| Metaproteómica | Metaproteoma: conjunto de proteínas expresadas por todos los miembros de comunidad en un determinado momento |
Para más información sobre estos y otros términos relacionados, se recomienda visitar la página web: http://www.genomicglossaries.com/content/omes.asp
El desarrollo a gran escala de las técnicas de la genómica y posgenómica ofrece una oportunidad sin precedentes para investigar los caracteres asociados a la patogenicidad de las especies fúngicas3. Además, existe una gran expectación sobre la posibilidad de aplicar dichos avances al diagnóstico de laboratorio de las micosis y al diseño de nuevas estrategias terapéuticas más eficaces.
Hoy día, el micólogo clínico ya dispone de una amplia variedad de técnicas de diagnóstico molecular, cada una de las cuales es útil para determinar la identidad del agente etiológico en una serie de niveles o rangos taxonómicos, pero pierde efectividad en otros aspectos. Es importante elegir bien la técnica que utilizar en función del uso que vayamos a hacer de ella: diagnóstico, epidemiológico o, simplemente, taxonómico.
Las técnicas moleculares para la detección e identificación de hongos patógenos pueden dividirse en dos tipos4:
- a)
Métodos de amplificación de señal, los cuales utilizan técnicas de hibridación de ácidos nucleicos. Las sondas de hibridación pueden utilizarse tanto para confirmar la identificación de un cultivo como para identificar el hongo patógeno en secciones tisulares. Estas sondas son utilizadas habitualmente para la identificación de patógenos como Histoplasma capsulatum y Coccidioides immitis.
- b)
Métodos de amplificación de ácidos nucleicos, que se refieren fundamentalmente a todas las técnicas de reacción en cadena de la polimerasa (PCR). Hay desarrolladas técnicas de PCR para el diagnóstico de un gran número de micosis; por ejemplo, en nuestro laboratorio aplicamos de forma habitual estas técnicas para el diagnóstico de la aspergilosis y las dermatofitosis animales5,6.
Por otro lado, debe considerarse el amplio abanico de técnicas disponibles para la tipificación molecular de los aislamientos fúngicos: técnicas basadas en la utilización de endonucleasas de restricción, en la amplificación de secuencias al azar, en la detección de secuencias cortas repetidas en tándem a lo largo del genoma (minisatélites y microsatélites), análisis de los polimorfismos de base única, etc. Las técnicas de tipificación molecular suelen aplicarse, fundamentalmente, al estudio epidemiológico de una infección y a la búsqueda de la fuente de contaminación por un patógeno. Sin embargo, la tipificación de cepas también es una herramienta útil para investigar la relación entre aislamientos recuperados de distintos pacientes y establecer la identidad de múltiples aislamientos de un mismo paciente7.
La investigación micológica en la era de la genómicaLo que parece más o menos claro es que tanto el diagnóstico como la investigación micológica se dirigen hacia una nueva era8, y existen al menos tres razones para explicar este hecho9:
- 1.
Las infecciones fúngicas empiezan a cobrar importancia como causa de mortalidad en los países desarrollados. A modo de ejemplo, Candida spp. es el cuarto microorganismo que, con más frecuencia, se aísla en sangre de pacientes estadounidenses.
- 2.
La industria de los antifúngicos tiene una gran importancia económica para las empresas farmacéuticas. Además, hay que tener en cuenta la aparición de resistencias y la necesidad de conocer su base genética.
- 4.
Se dispone ya de la secuencia de los genomas de los principales patógenos fúngicos (Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans) y se está trabajando en la secuenciación de otros muchos (Candida tropicalis, Pneumocystis jiroveci, Histoplasma capsulatum).
Tampoco debemos obviar otros factores que han contribuido a esta nueva concepción, como son la mejora en las tecnologías de secuenciación y análisis de datos, que permiten la reducción de costes económicos y esfuerzo humano, y la disponibilidad de patrocinadores dispuestos a sostener económicamente la investigación genómica en hongos una vez que el Proyecto Genoma Humano (PGH) se encuentra en su fase final8. Por todo ello, hay quien considera que la genómica, es decir, el estudio de toda la información genética contenida en un organismo, va a tener un papel preponderante durante los próximos años.
Algunos hitos en la genómica de los hongosLa historia de la genómica en hongos es corta pero intensa. Esta carrera por descifrar los secretos que esconde el genoma de los hongos comenzó en 1992, con la publicación de la secuencia completa del cromosoma III de Saccharomyces cerevisiae.10 En 1996, 4 años más tarde, se hizo pública la secuencia completa de todo el genoma de esta especie de levadura11. Estos dos eventos son considerados de gran relevancia no sólo por los micólogos, sino también por genéticos y biólogos celulares, pues representan el primer cromosoma y el primer genoma eucariota en secuenciarse.
Tras la gran expectación inicial, se empezó a discutir sobre la necesidad de dar un paso más y abordar en profundidad el estudio de los genomas de hongos filamentosos.
De estas discusiones surgieron pequeños proyectos piloto de secuenciación de partes de genomas como fue, por ejemplo, el proyecto de secuenciación del cromosoma IV de Aspergillus nidulans. Pero no fue hasta el año 2000 cuando la genómica de los hongos filamentosos comenzó a ver la luz, con el proyecto denominado Fungal Genome Initiative (FGI). En la gestación de esta iniciativa participaron distintos miembros de la comunidad científica y consorcios de secuenciación, de entre los que destaca el Whitehead Institute for Biomedical Research (Cambridge, Massachusetts, EE.UU.), conocido en la actualidad como Broad Institute. Finalmente, en 2003 se hizo pública la secuencia completa del genoma de Neurospora crassa,12 el primero correspondiente a un hongo filamentoso.
The Fungal Genome InitiativeSin lugar a dudas, el gran impulso para la genómica de los hongos filamentosos ha sido el proyecto denominado Fungal Genome Initiative (FGI). La filosofía y las principales líneas de este proyecto están recogidas en cuatro documentos13–16. Además, desde la gestación de esta iniciativa en el año 2000 se han ido sumando distintas instituciones y consorcios de secuenciación de genomas. De entre todas estas instituciones, destaca la participación del Broad Institute, por actuar como "centro neurálgico" de este proyecto y por haber colaborado activamente en la secuenciación de algunos de los genomas que han despertado mayor interés (A. nidulans, A. terreus, C. albicans, C. immitis, C. neoformans). Es importante tener en cuenta que en un proyecto de tal magnitud resulta indispensable que todas las instituciones implicadas estén perfectamente coordinadas.
En la propuesta inicial de este gran proyecto13 se incluyeron cinco especies de hongos con importancia para la salud humana (C. neoformans serotipo A, Coccidioides posadasii, P. jiroveci, Trichophyton rubrum y Rhizopus oryzae), además de especies con importancia económica o como organismos modelo. Las especies fúngicas incluidas en el proyecto fueron elegidas siguiendo una serie de criterios13: importancia del organismo para el ser humano (impacto sobre la salud o la economía); valor del organismo como herramienta para los estudios de genómica comparada; existencia de recursos genéticos para llevar a cabo el proyecto; interés de la comunidad científica en el proyecto (existencia de grupos de trabajo establecidos).
La gran aportación de esta iniciativa ha sido afrontar los proyectos genoma de hongos desde una perspectiva taxonómica8,13, por considerarse que una selección equilibrada de los organismos fúngicos que van a ser secuenciados maximizaría el valor de los datos obtenidos, al permitir estudios evolutivos y de genómica comparada. Esto contrasta con los pequeños proyectos independientes anteriores a esta gran iniciativa.
Situación actual de los proyectos genoma en hongosHoy en día se conoce la secuencia de más de 18 especies de hongos. En líneas generales, se puede decir que mientras que las levaduras centraron toda la atención durante los primeros años de la genómica en hongos, ahora se ha invertido la tendencia y la mayoría de los proyectos de secuenciación en curso se centran en los hongos filamentosos8.
También está empezando a cobrar cierta importancia otro tipo de proyectos que no pretenden una secuenciación completa de los genomas fúngicos, sino la identificación de los genes que se expresan en un organismo en un momento determinado8,17. A estos fragmentos de ADN se les conoce con el nombre de etiquetas de secuencia expresada o expressed sequence tags (EST) y la principal ventaja de este abordaje es la posibilidad de identificar las regiones codificantes del genoma que se expresan bajo determinadas condiciones. Por ejemplo, mediante esta aproximación podemos identificar los genes que expresa un hongo patógeno que se encuentra en el interior de un hospedador, entre los cuales habrá posibles factores de virulencia.
Genómica funcionalLa secuenciación genómica de microorganismos se ha convertido en una operación casi sistemática y no representa en sí misma un desafío18. Lo que presenta una mayor complejidad es interpretar la información obtenida de la secuenciación de un genoma. Según Foster et al8, la secuencia de un genoma es similar a un diccionario que recoge todas las palabras pero no incluye las definiciones. A partir de esa secuencia, se trata de buscar marcos abiertos de lectura (open reading frames [ORF]), identificar la función o funciones de cada gen y las posibles interacciones entre ellos. En líneas generales, resulta más difícil identificar ORF en hongos filamentosos que en levaduras, no sólo por la mayor complejidad de la estructura de sus genes, sino también por conocerse menos acerca de los mecanismos de regulación de su expresión8.
A continuación describimos brevemente las dos formas básicas de abordar la genómica funcional. La primera de ellas consiste en analizar los patrones de expresión génica, para lo cual suele recurrirse a los chips de ADN. La otra forma de abordar la genómica funcional es analizar directamente la función de los genes anulando ésta por mutación.
Los arrays de ADNUn array (matriz) es un conjunto de sondas moleculares fijadas de manera ordenada sobre un soporte sólido19. Cuando estas sondas son moléculas de ADN se habla de arrays de ADN. Esta técnica se basa en el principio de hibridación específica entre un ARN mensajero (ARNm) y la molécula de ADN de la cual deriva por transcripción. Se puede consultar una revisión detallada sobre los arrays en un número anterior de esta misma revista19.
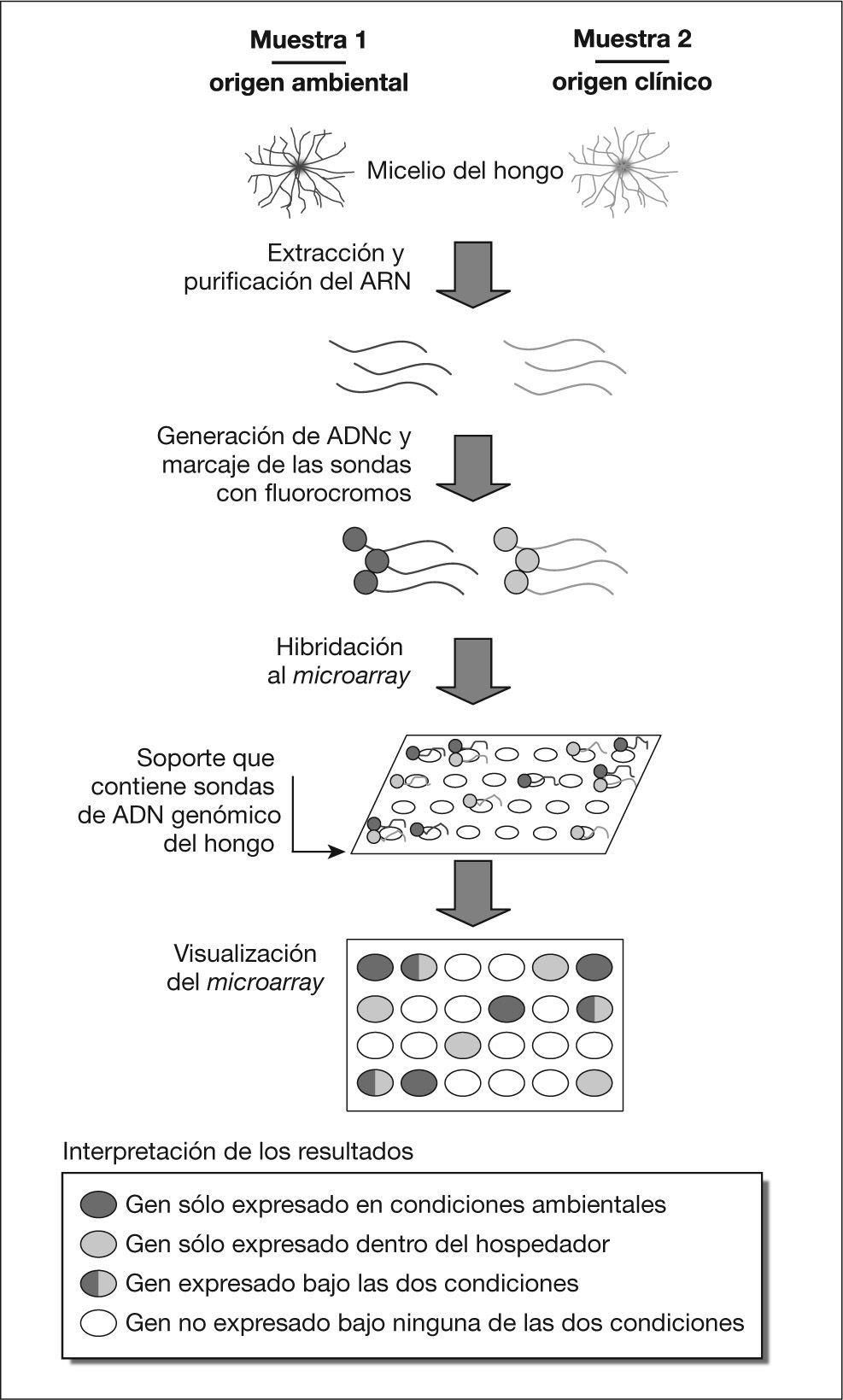
La tecnología de arrays o chips de ADN se utiliza cada vez con más frecuencia para estudiar simultáneamente y de manera cuantitativa la expresión de genes a escala genómica, sobre todo desde que se está intentando reducir el coste y la complejidad de la técnica a la vez que se estandariza. En el caso de los estudios de patogenicidad, la idea subyacente es que la identificación de los patrones de expresión génica del hongo patógeno bajo dos condiciones totalmente diferentes, como pueden ser las condiciones ambientales y las asociadas a una infección, puede ayudar en la búsqueda de potenciales factores de virulencia20. Este proceso de búsqueda de genes relacionados con la patogenicidad fúngica se esquematiza en la figura 1.

A modo de ejemplo, se considera que el estudio de la expresión génica en Aspergillus fumigatus durante los procesos infecciosos mediante el uso de chips de ADN permitirá, en un futuro no muy lejano, ampliar los conocimientos sobre la virulencia de este hongo21. Un ejemplo de este tipo de estudios se refiere a la termotolerancia de A. fumigatus, característica que es considerada un factor esencial para su crecimiento dentro del hospedador y en algunos ambientes naturales en los que dominan las temperaturas elevadas. Utilizando la técnica de microarrays de ADN, Nierman et al22 examinaron los perfiles de expresión génica en A. fumigatus a tres temperaturas diferentes: 30, 37 y 48 °C, temperaturas que representan los ambientes de un suelo tropical, el cuerpo humano y una pila de compost, respectivamente22,23. El microarray utilizado fue construido utilizando amplicones obtenidos por PCR para cada uno de los genes del genoma. Entre los interesantes resultados obtenidos por estos autores se encuentra la observación de que a 37 °C se expresa un mayor número de genes relacionados con la patogenicidad (p. ej., genes que codifican proteínas que responden al estrés oxidativo o al sistema inmunitario del hospedador y genes implicados en la producción de toxinas) que a las otras dos temperaturas probadas, aunque no haya contacto alguno con células de mamíferos o productos de éstas23.
El análisis de mutantesEl análisis de mutantes de deleción para inferir la función de los genes y realizar cartografías cromosómicas tiene ya bastante tradición en genética. Este tipo de análisis solía hacerse de manera individual, es decir, utilizando un único mutante en cada ensayo. Sin embargo, en la era de la genómica se pretende obtener y analizar mutantes de forma masiva. Tras generar las colecciones de mutantes, por ejemplo por transposición, hay que realizar un cribado para encontrar los microorganismos que sean incapaces de realizar alguna función particular o carezcan de un determinado factor de patogenicidad.
Un método que se presenta como una alternativa atractiva al análisis de mutantes de manera individual es la generación de mutantes "etiquetados", más conocido como signature-tagged mutagenesis (STM)24. Esta técnica, desarrollada por primera vez en Salmonella para la identificación de genes imprescindibles para el crecimiento bacteriano in vivo, consiste en la creación de una colección de cepas mutantes que contengan "etiquetas" identificativas únicas (a modo de códigos de barras moleculares), es decir, secuencias específicas que permitan identificar a cada uno de los miembros de la colección. La colección de cepas construida es utilizada para infectar a un determinado hospedador. La posterior comparación de la abundancia relativa de cada cepa individual en el conjunto que se introduce al hospedador y en el que se obtiene de éste, permite identificar los genes requeridos para el crecimiento in vivo y/o la virulencia25.
La aplicación de esta técnica en la identificación de genes fúngicos requeridos para el crecimiento in vivo se enfrenta a varias dificultades. Entre estas dificultades destacan la gran cantidad de regiones no codificantes existente en los genomas fúngicos, los cuales, además, suelen ser de gran tamaño, y la falta de métodos eficaces de mutagénesis insercional26. No obstante, Brown et al26 consiguieron adaptar dicha técnica para identificar los genes requeridos para el crecimiento in vivo de A. fumigatus. De esta forma, lograron obtener una cepa incapaz de causar infección letal en un modelo animal, la cual resultó ser un mutante con una inserción en el promotor del gen pabaA. Éste codifica para la enzima ácido paraaminobenzoico (PABA) sintetasa, la cual interviene en uno de los últimos pasos de la biosíntesis del folato, cofactor esencial de la síntesis del ADN. Al parecer, el pulmón del hospedador no proporciona este compuesto al hongo21, por lo que esta enzima se considera esencial para el crecimiento de A. fumigatus en el pulmón26,27.
Cuando se realiza un análisis de mutantes para identificar factores supuestamente relacionados con la patogenicidad de un microorganismo hay que tener ciertas precauciones. Una vez que se ha identificado un factor de virulencia potencial hay que recurrir a estudios de complementación para ver si se cumplen los denominados "postulados de Falkow" o "postulados moleculares de Koch"28. Según estos postulados, para poder ligar un gen a la patogenicidad de un microorganismo se requiere la concurrencia de los siguientes hechos:
- 1.
Que el fenotipo o propiedad bajo investigación esté asociado a especies patógenas de un mismo género o cepas patógenas de una misma especie, lo cual viene a indicar que un gen o factor de virulencia no va a ser exclusivo de una cepa o especie, sino una estrategia dentro de una especie o especies relacionadas.
- 2.
Que la inactivación específica del gen o genes asociados con el presunto carácter de virulencia conlleve una pérdida medible de la virulencia o la patogenicidad del microorganismo.
- 3.
Que la reversión o reemplazamiento alélico del gen mutado (reintroducción del gen del tipo salvaje) conlleve la restauración de la patogenicidad o virulencia perdida.
Estos postulados han sido comprobados para algunos factores de virulencia de agentes causantes de micosis sistémicas, como son los genes que codifican la cápsula de C. neoformans29.
Genómica comparadaLa genómica comparada, encargada del análisis y la comparación de los genomas de diferentes organismos, es una disciplina de gran utilidad. Para ilustrar este punto podemos poner el ejemplo del género Aspergillus. Dentro de este género, varias especies son utilizadas en procesos industriales (producción de ácidos, fármacos, enzimas, etc.). Por su parte, especies como A. fumigatus son patógenas oportunistas del hombre y otros animales, mientras que otros miembros del mismo género están implicados en la degradación de alimentos y la producción de metabolitos secundarios tóxicos (p. ej., producción de aflatoxinas por A. flavus). Finalmente, dentro de este género hay especies que sirven de modelo en genética y biología celular. Destacan en este campo las investigaciones con A. nidulans. La utilidad de la genómica comparada es evidente en casos como el de A. fumigatus, organismo del que se desconoce la función de la mitad de los genes encontrados en su genoma. Por comparación de secuencias con otras especies cercanas es posible llegar a atribuir funciones a algunos de esos genes. Además, comparar el genoma de especies patógenas con otras no patógenas relacionadas permite identificar secuencias que, potencialmente, contienen genes o elementos reguladores de la virulencia3. En este sentido, la disponibilidad de la secuencia genómica de diversas especies del género Aspergillus representa para la comunidad científica una oportunidad sin precedentes30.
Cabe destacar, igualmente, el hecho de que para algunas especies fúngicas, como es el caso de A. fumigatus, C. albicans y C. neoformans se está llevando a cabo la secuenciación de varias cepas20. Esta aproximación va a permitir realizar estudios comparativos de las diferencias genotípicas existentes en el seno de una misma especie, por ejemplo, entre cepas patógenas y no patógenas o entre diferentes serotipos.
La genómica comparada permite, además, diseñar sondas de ADN que sirvan para la identificación rápida y precisa de la especie. Esta utilidad es de gran interés para la micología médica, sobre todo si se tiene en cuenta que el tratamiento de algunas micosis es diferente según el agente causal sea una especie u otra, debido a la aparición de fenómenos de resistencia a los antifúngicos. Otra aplicación práctica de la genómica comparada es ayudar a resolver la taxonomía de algunos grupos de hongos. Así, por ejemplo, el estudio de la secuencia del gen CHS1, que codifica para la enzima quitina sintasa 1, ha servido para analizar las relaciones filogenéticas entre diversas especies de dermatofitos31–33. Estos estudios moleculares han servido como complemento a las otras técnicas utilizadas tradicionalmente en la taxonomía fúngica, las cuales se basaban en caracteres morfológicos y bioquímicos31.
Por último, dentro de la genómica comparada se sitúa el estudio de diversos marcadores moleculares, entre ellos los polimorfismos de base única (single nucleotide polymorphisms [SNP]). Los SNP son sitios específicos dentro de un genoma en los cuales diferentes individuos pueden presentar una base nucleotídica distinta. Los SNP pueden localizarse en las regiones codificantes del genoma, llegando incluso a alterar la estructura y función de las proteínas codificadas, pero, sobre todo, se distribuyen en las regiones intergénicas. Forche et al34 describieron, al cartografiar la presencia de polimorfismos de base única en el genoma de C. albicans, la existencia de 561 SNP en el genoma de esta especie de levadura. Estos polimorfismos, que en C. albicans se presentan distribuidos con una frecuencia media de 1 SNP por cada 83 pares de bases, pueden utilizarse como marcadores a la hora de establecer la relación genética entre individuos, además de ayudar en el estudio de procesos tales como la recombinación y la ocurrencia de reordenaciones en el genoma34.
ProteómicaEl término "proteoma" fue acuñado en la década de 1990 para describir el conjunto de proteínas de una célula, tejido u organismo35,36. Por tanto, la proteómica es la disciplina científica encargada de desarrollar la tecnología necesaria para analizar los patrones globales de expresión de proteínas en un organismo. Se reconoce la existencia de al menos tres factores decisivos para el desarrollo de la proteómica36:
- 1.
Los proyectos de secuenciación de genomas a gran escala y el desarrollo de bases de datos de proteínas.
- 2.
El desarrollo de técnicas de espectrometría de masas para analizar proteínas y péptidos.
- 3.
Los avances en las técnicas de separación de proteínas.
La diferencia básica entre genómica y proteómica es que mientras que la primera engloba el estudio de todo el patrimonio genético de un organismo (tanto nuclear como extranuclear, codificante o no codificante), la segunda se centra en las partes del genoma que se traducen a proteínas. Estas dos disciplinas también difieren en su naturaleza. A diferencia del genoma, que permanece relativamente estático, el proteoma es dinámico35,36. El conjunto de proteínas que se expresan no sólo varía de una célula a otra, sino que depende de las interacciones entre el genoma y el ambiente en un momento determinado18, de manera que cualquier genoma puede, potencialmente, dar lugar a un número infinito de proteomas35,36.
Lo que resulta más interesante de la proteómica es que permite abordar las características biológicas que no pueden ser identificadas mediante el análisis en exclusiva del ADN3,35: abundancia relativa de una proteína, modificaciones postraduccionales, localización subcelular, tasa de renovación, posibles interacciones con otras proteínas, etc. Además, la proteómica constituye una herramienta fundamental para la "anotación" de un genoma, es decir, para asignar a cada gen su función. Pero, sin duda, el objetivo último de la proteómica es obtener una visión global e integrada de todos los procesos celulares36.
Al igual que en el caso de la genómica, existe una tendencia a subdividir la proteómica en varias disciplinas. Concretamente, se suelen diferenciar los siguientes tipos35,36:
- –
Proteómica de expresión: es el estudio cuantitativo de los patrones de expresión proteica entre muestras que se diferencian en alguna variable.
- –
Proteómica estructural o del mapa celular: se encarga del estudio de la localización subcelular de las proteínas, así como de las interacciones proteína-proteína.
- –
Proteómica funcional: se refiere al estudio y la caracterización de un grupo de proteínas determinado.
Por otro lado, en la metodología de una investigación proteómica suelen diferenciarse tres fases35: a) el aislamiento y la separación de proteínas a partir de una muestra biológica; b) la obtención de información sobre la estructura de las proteínas separadas, y c) la utilización de bases de datos informáticas para poder identificar las proteínas caracterizadas.
Para el aislamiento y la separación de proteínas se utilizan, básicamente, técnicas electroforéticas. El desarrollo de la electroforesis bidimensional en geles de poliacrilamida (2D-PAGE), basada en una separación de las proteínas en función de la carga seguida de una separación en función de su masa molecular, constituyó un gran avance en el campo de la proteómica35. No obstante, esta técnica presenta ciertas limitaciones35,36: es laboriosa y difícil de automatizar, está limitada por el número y el tipo de proteínas que se pueden resolver y, sobre todo, utilizando esta técnica se identifican predominantemente proteínas abundantes, mientras que las proteínas que muestran una baja abundancia no suelen ser detectadas. En cuanto a las técnicas empleadas para obtener información sobre la estructura de las proteínas, destacan la secuenciación de los extremos amino y carboxilo terminal, y la espectrometría de masas (MS).
Además de las técnicas ya mencionadas, cabe destacar la aparición de los primeros microarrays de proteínas, los cuales presentan un enorme potencial no sólo en la investigación básica, sino también en el diagnóstico y en la búsqueda de nuevos agentes terapéuticos37,38. A diferencia de los ácidos nucleicos, las proteínas suelen ser más difíciles de preparar para el formato de un microarray38, ya que para que éstas sean biológicamente activas se requiere que su adherencia al soporte no suponga una pérdida de la conformación original. En los arrays de proteínas, al igual que en el caso de los arrays de ADN, existe cierta diversidad en cuanto al tipo de soporte que se puede utilizar. Aunque se pueden fabricar arrays de proteínas en los que el soporte sea una membrana de nitrocelulosa o un portaobjetos de vidrio, estos materiales suelen sustituirse por matrices tridimensionales de acrilamida o agarosa. Estas matrices tienen una estructura muy porosa e hidrofílica, lo cual contribuye a mantener un ambiente húmedo que evita la desnaturalización y ayuda a conservar el estado activo de las proteínas38. También se han desarrollado estructuras abiertas, consistentes en chips de 18 x 28 mm que incluyen diminutos pocillos (1,4 mm de diámetro y 300 ^m de fondo) y están fabricados a partir de un elastómero de silicona, el polidimetilsiloxano (PDMS)39. Estos chips pueden disponerse sobre un cubreobjetos de vidrio para facilitar su manejo. También existen diferentes métodos para unir las proteínas a la superficie del soporte, desde la simple adsorción pasiva hasta las interacciones de alta afinidad38. Por último, también hay que considerar la enorme diversidad de métodos de detección de señal, ya sea de reacción con la proteína del soporte o de unión a ésta. Aunque suelen preferirse los métodos de detección de fluorescencia, a partir de su sencillez y extremada sensibilidad, también se puede utilizar un marcaje con radioisótopos, métodos de detección basados en reacciones enzimáticas, etc.38,40.
La mayor versatilidad de los arrays de proteínas respecto de los arrays de ADN también queda de manifiesto si tenemos en cuenta los tipos de proteínas que pueden adherirse al soporte inerte para utilizarse como sondas. Así, por ejemplo, se puede hablar de microarrays de anticuerpos, microarrays de antígenos y microarrays de enzimas, según el tipo de sonda que se utilice (anticuerpos, antígenos y enzimas, respectivamente). Del mismo modo, también se pueden fabricar microarrays de péptidos. Esta característica hace que los arrays de proteínas sean una herramienta ideal para analizar la función de todo tipo de proteínas y las interacciones moleculares en las que éstas participan.
Hasta la fecha, el análisis del proteoma de los hongos se ha centrado en estudiar la estructura y composición de su pared celular, buscar y caracterizar potenciales factores de virulencia, intentar entender el proceso de dimorfismo fase micelial-fase de levadura que presentan muchos hongos patógenos y estudiar la base molecular de la resistencia a los antifúngicos3. Respecto a este último aspecto, Hooshdaran et al41 realizaron un estudio de los patrones de expresión proteica en dos cepas de C. albicans, una sensible y otra resistente al fluconazol. Para ello utilizaron la técnica de electroforesis bidimensional (2D-PAGE). En la cepa resistente encontraron 13 proteínas sobreexpresadas y cuatro reprimidas con respecto a la cepa sensible. Entre las proteínas que mostraron una sobreexpresión en la cepa resistente destacan diversas enzimas involucradas en el metabolismo de los carbohidratos y Erg10p, proteína involucrada en el primer paso de ruta biosintética del ergosterol. Estos resultados sirvieron para confirmar observaciones anteriores en referencia a la sobreexpresión de genes involucrados en la biosíntesis del ergosterol, entre ellos el gen ERB10, en respuesta al tratamiento con itraconazol42.
Bioinformática y nanotecnologíaEl desarrollo de la genómica y de lo que se ha venido a denominar como posgenómica no habría sido posible sin la aparición de una nueva disciplina: la bioinformática. Como su propio nombre indica, la bioinformática es un área de investigación posicionada en la interfase entre dos ciencias: la informática y la biología.
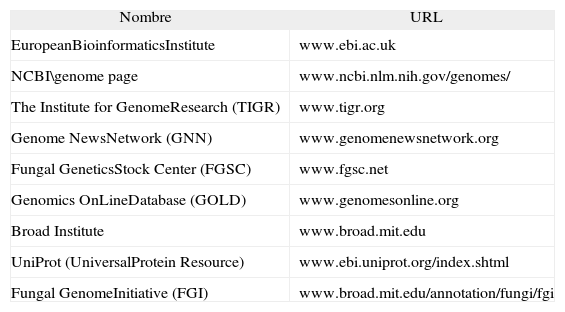
El desarrollo de la bioinformática debe entenderse en el marco de la llamada "revolución genómica" y del rápido desarrollo de las tecnologías de la información. La bioinformática surge como respuesta a la urgente necesidad de disponer de herramientas informáticas que permitiesen manejar, interpretar y distribuir el gran volumen de información obtenida en los proyectos genoma. Básicamente, esta disciplina pretende resolver problemas biológicos complejos utilizando las herramientas informáticas disponibles en la actualidad. Así pues, los bioinformáticos se encargan de desarrollar fórmulas y modelos matemáticos que asistan a los investigadores en la comparación de secuencias, búsqueda de ORF, estudio conformacional de las biomoléculas y resolución de filogenias. Por otro lado, internet está siendo un instrumento básico para el manejo y la distribución de toda la información relativa a los proyectos de genómica y proteómica. En la tabla 2 se presentan, a modo de ejemplo, algunos sitios web de interés relacionados con la genómica, la proteómica y la bioinformática.
Algunos sitios web de interés relacionados con la genómica, la proteómica y la bioinformática
| Nombre | URL |
| EuropeanBioinformaticsInstitute | www.ebi.ac.uk |
| NCBI\genome page | www.ncbi.nlm.nih.gov/genomes/ |
| The Institute for GenomeResearch (TIGR) | www.tigr.org |
| Genome NewsNetwork (GNN) | www.genomenewsnetwork.org |
| Fungal GeneticsStock Center (FGSC) | www.fgsc.net |
| Genomics OnLineDatabase (GOLD) | www.genomesonline.org |
| Broad Institute | www.broad.mit.edu |
| UniProt (UniversalProtein Resource) | www.ebi.uniprot.org/index.shtml |
| Fungal GenomeInitiative (FGI) | www.broad.mit.edu/annotation/fungi/fgi |
Para llevar a cabo sus objetivos, la bioinformática dispone de una serie de bases de datos y distintos paquetes de software. De entre todas las bases de datos disponibles destaca el GenBank. El GenBank es una colección anotada de todas las secuencias de nucleótidos y aminoácidos disponibles al público gestionada por el National Center for Biotechnology Information (NCBI) de Estados Unidos. El NCBI fue creado en 1988 para desarrollar herramientas bioinformáticas que sirviesen para almacenar y analizar todo el conocimiento sobre biología molecular, bioquímica y genética. En sus orígenes, era el personal del NCBI el encargado de buscar en la literatura científica todas las secuencias de ácidos nucleicos y aminoácidos e incluirlas en el GenBank. Hoy día, sin embargo, casi todas las secuencias son depositadas directamente en el GenBank por los laboratorios que las generan. Esto se debe, al menos en parte, al hecho de que la mayoría de las revistas ponen como condición a los autores que las secuencias que aparecen en sus trabajos de investigación sean depositadas previamente en una base de datos accesible al público43.
Entre las bases de datos sobre información proteómica destaca UniProt (Universal Protein Resource), en la cual participan diversas instituciones. UniProt contiene un completo catálogo de proteínas en el que no sólo se incluye información sobre su estructura y función, sino también sobre la localización subcelular de cada proteína, la familia a la que ésta pertenece, sus posibles actividades catalíticas, etc.
Respecto a los paquetes de software bioinformático disponibles, son ya famosos algunos programas para el análisis filogenético a partir de secuencias de nucleótidos y aminoácidos, como son los programas PAUP (Phylogenetic Analysis Using Parsimony) y PHYLIP (PHYLogeny Inference Package). El programa PAUP, en sus diferentes versiones, es el paquete comercial más utilizado en la realización de este tipo de análisis. Por su parte, PHYLIP es un paquete informático que incluye diversos programas para inferir filogenias y construir árboles evolutivos a partir de las secuencias de ADN y de proteínas. El acceso a este último software puede realizarse de manera gratuita a través de internet (http://evolution.genetics.washington. edu/phylip.html).
No debemos concluir este apartado sin considerar las importantes contribuciones de la nanotecnología en el campo de la genómica y la proteómica. Así, por ejemplo, la fabricación de chips de ácidos nucleicos y proteínas ha sido posible gracias al desarrollo parejo de las tecnologías de micromanipulación y fotolitografía, que permiten, respectivamente, la deposición de microgotas que contengan las sondas que testar sobre el soporte inerte y la síntesis directa de la sondas sobre dicho soporte.
Algunas aplicaciones prácticas de la genómica y la proteómica en el diagnóstico micológicoEntre los argumentos para impulsar los proyectos de secuenciación en hongos se encuentra la necesidad de disponer de mejores métodos de diagnóstico y de nuevos agentes terapeúticos44.
Leinberger et al2 han sido los primeros en describir la aplicación de la tecnología de microarrays de ADN para el diagnóstico rápido de las 12 especies patógenas más comunes de los géneros Aspergillus y Candida, utilizando para ello sondas de captura diseñadas a partir de la secuencia de las regiones espaciadoras internas (ITS) de los genes que codifican para el ARN ribosómico. Estas regiones de ADN son particularmente útiles en este sentido, debido a que2:
- 1.
Presentan numerosas copias en el genoma fúngico.
- 2.
Se dispone de primers universales, válidos para cualquier especie fúngica, basados en las regiones conservadas de los genes que codifican para el ARNr.
- 3.
Las regiones ITS presentan un grado de variabilidad suficiente como para permitir la distinción de la especie.
Entre los objetivos para el futuro se encuentra el diseño de un microarray que permita identificar la presencia de un mayor número de patógenos fúngicos en muestras clínicas2.
La genómica fúngica se encuentra en posición de desempeñar un papel fundamental en el descubrimiento de nuevos antifúngicos25. La necesidad de desarrollar nuevos fármacos para tratar las infecciones fúngicas puede justificarse en los siguientes puntos25: las limitaciones terapéuticas de los antifúngicos ya existentes, como los polienos, que presentan problemas de toxicidad, o los azoles, que muestran un limitado espectro de eficacia; el incremento en la incidencia de las infecciones fúngicas y la emergencia de aislamientos resistentes. Los productos de los genes esenciales de algunos hongos como A. fumigatus son considerados dianas potenciales para los fármacos antifúngicos22,44. Sin embargo, debe tenerse en cuenta que el fenotipo que muestra un organismo es el resultado de la interacción entre su genotipo y el ambiente, por lo que sólo tiene sentido hablar de genes esenciales con relación a unas condiciones ambientales determinadas45.
Los métodos moleculares presentan también un enorme potencial a la hora de evaluar la susceptibilidad a los antifúngicos, sobre todo en el caso de hongos difíciles de cultivar. Entre las ventajas que ofrece la detección molecular de la resistencia a antifúngicos, destaca la posibilidad de examinar directamente muestras clínicas, sin tener que realizar un aislamiento previo del hongo46. Además, al partir de una muestra clínica estaremos analizando en su conjunto una población del hongo, en lugar de una única colonia seleccionada al azar a partir de un cultivo46. En este sentido, López-Ribot et al47 observaron, al estudiar episodios de candidiasis orofaríngea en pacientes infectados por el VIH, que aislamientos clínicos obtenidos del mismo paciente pueden presentar heterogeneidad en sus patrones de expresión de los genes involucrados en la resistencia al fluconazol. Además, en la misma etapa del proceso infectivo pueden coexistir dentro de un mismo paciente diferentes subpoblaciones de levaduras, las cuales pueden desarrollar la resistencia al antifúngico por mecanismos diferentes47. Si seguimos los procedimientos habituales de determinación de la susceptibilidad, basados en la determinación del crecimiento o actividad metabólica en medios que contengan diferentes concentraciones del antifúngico en prueba, estaremos perdiendo una información valiosísima: el mecanismo molecular por el cual aparece dicha resistencia.
Por último, los microarrays de proteínas se postulan como una interesante alternativa para el cribado de sustancias fúngicas capaces de desencadenar una respuesta inmune. Así, por ejemplo, Lebrun et al40 han logrado combinar la sensibilidad de los ensayos tipo ELISA con las múltiples ventajas de la tecnología de miroarrays para crear un sistema capaz de detectar pequeñas cantidades de inmunoglobulina E (IgE) producidas en respuesta a alérgenos de origen fúngico. Lo interesante de esta nueva aproximación al diagnóstico de las hipersensibilidades de tipo I es que permite identificar, para cada paciente, el componente fúngico causante de la enfermedad de entre todos los presentes en un extracto alergénico.
ConclusionesEn los últimos tiempos estamos asistiendo a una generalización en el uso de las mismas técnicas moleculares que hasta hace pocos años sólo estaban disponibles en los laboratorios más avanzados, lo que, sin duda, constituye un gran avance. Todos los esfuerzos por maximizar la eficiencia y minimizar el coste de aplicación de estas técnicas serán, sin duda, recompensados por la gran cantidad de nueva información sobre la base molecular de la patogénesis fúngica a la que tendremos acceso.
Entre los desafíos futuros de la genómica y la proteómica en el campo de la investigación micológica se encuentra la posibilidad de identificar genes que codifican factores de patogenicidad en los principales agentes causantes de micosis y estudiar su expresión en diferentes estadios del proceso infeccioso. También debería priorizarse la aplicación de las técnicas que ofrecen estas nuevas disciplinas al estudio de la respuesta del huésped al patógeno fúngico.
Aunque todavía queda un largo camino para que estas tecnologías de nueva generación se pongan al servicio del diagnóstico de laboratorio, el micólogo clínico no debería ser ajeno a los rápidos avances en el campo de la genómica y la proteómica, pues sólo así estará en disposición de aprovechar al máximo las oportunidades que esta revolución científica nos ofrece.

