




Rilpivirina (RPV) es un nuevo inhibidor de la transcriptasa inversa no análogo de nucleósidos de segunda generación, aprobado recientemente para el tratamiento de pacientes con infección por el virus de la inmunodeficiencia humana a dosis de 25mg, administrado 1 vez al día con alimentos. En pacientes que inician tratamiento, RPV se ha evaluado en 1 estudio fase IIb (TMC278-C204) y en 3 estudios fase III (ECHO, THRIVE y STaR). En todos ellos, RPV se compara con el gold standard (efavirenz [EFV]), incluyendo un considerable número de pacientes tratados con el nuevo fármaco (n=1.349). La eficacia terapéutica ha resultado elevada y similar con ambos tratamientos, demostrándose que RPV es no inferior a EFV a las 48 y 96 semanas. En todos los estudios y en todos los subgrupos analizados, RPV ha mostrado una mejor tolerabilidad que EFV, especialmente en lo referido a efectos adversos del área neuropsiquiátrica, exantema cutáneo y de la esfera lipídica. En el análisis conjunto de los estudios ECHO y THRIVE se observaron diferencias notables en función de la carga viral (CV) basal. Así, la eficacia terapéutica de RPV resultó superior a la de EFV en pacientes con CV basal ≤ 100.000 copias/ml, con similar eficacia virológica y, además, una mejor tolerabilidad de RPV. Por el contrario, en los pacientes con CV basal ≥ 100.000 copias/ml se observó una mayor incidencia de fallos virológicos en el grupo de RPV, especialmente con CV basal ≥ 500.000 copias/ml. La aparición de mutaciones de resistencia en los pacientes con fallo virológico resultó superior a la esperada, especialmente en los pacientes con CV basal elevada. Según estos resultados, las agencias del medicamento europeo (EMA) y americana (FDA) han aprobado el uso de RPV en pacientes naïve con CV basal ≤ 100.000 copias/ml. Algunas guías terapéuticas ya incorporan RPV en sus recomendaciones. Así, las americanas (DHHS e IAS-USA), en espera de datos adicionales, la consideran todavía como un régimen alternativo, mientras que las europeas y la reciente actualización de las Guías de GeSIDA la consideran entre los regímenes preferidos en pacientes con CV ≤ 100.000 copias/ml. Los datos recientes del estudio STaR, en el que se utilizan combinaciones a dosis fijas de los 3 fármacos, muestran no inferioridad de RPV frente a EFV, independientemente de la CV basal, con menos fallos virológicos y una menor selección de resistencias que en los estudios ECHO y THRIVE. En definitiva, los datos de eficacia y seguridad del fármaco indican que los regímenes basados en RPV con 2 inhibidores de la transcriptasa inversa análogos de nucleósidos son eficaces y seguros en el tratamiento antirretroviral de inicio.
Rilpivirine (RPV) is a new, second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) that has been recently approved for use in the initial antiretroviral therapy (ART) of treatment-naïve HIV-infected patients, combined with two nucleoside/nucleotide reverse transcriptase inhibitors (NRTI). The approved dose is 25mg once daily with food. RPV has been assessed in a phase IIb study (TMC278-C204) and in three phase III trials (ECHO, THRIVE and STaR). In all of them, RPV was compared with the gold standard, efavirenz (EFV); these studies enrolled a large number of patients (n=1,349 on RPV). RPV was non-inferior to EFV at 48 and 96 weeks. In all the studies and study arms, the tolerability of RPV was better than that of EFV, especially for neuropsychiatric adverse effects, rash, and lipid profile. An analysis of the combined data from the ECHO and THRIVE trials showed marked differences, depending on baseline viral load. The therapeutic efficacy of RPV was superior to that of EFV in patients with a baseline viral load ≤100,000 copies/mL, due to a similar virological efficacy and a better tolerability profile. However, in patients with a baseline viral load ≥100,000 copies/mL, virological failure was more frequent in the RPV arm, especially in patients with a viral load ≥500,000 copies/mL. Emerging resistance mutations to RPV were commonly detected in patients with virological failure, especially in those with a higher baseline viral load. In view of these results, the European Medications Agency and the US Food and Drug Administration have approved the use of RPV in treatment-naïve patients with a baseline viral load ≤100,000 copies/mL. Some treatment guidelines have already included RPV among their recommendations. The guidelines of the US Department of Health and Human Services (DHSS) and the International Antiviral Society-USA ((IAS-USA), while awaiting additional data, consider RPV-based regimens as an alternative regimen. The Gesida guidelines consider RPV to be among the preferred regimens in patients with a viral load ≤100,000 copies/mL. Recent data from the STaR trial, which used fixed drug combinations, have shown the non-inferiority of RPV with respect to EFV, less virological failure and less emergence of resistance mutations with RPV use, irrespective of baseline viral load. In summary, efficacy and safety data suggest that RPV plus 2 NRTI is an effective and safe initial antiretroviral regime.
Hace ya más de 30 años que se inició la epidemia de infección por el virus de la inmunodeficiencia humana (VIH) y más de 20 años que se utilizan fármacos antirretrovirales (ARV) para su tratamiento. Los objetivos del tratamiento frente a la infección por el VIH han ido cambiando progresivamente, al mismo tiempo que se lograban importantísimos avances en el conocimiento de la infección, del virus y del propio tratamiento ARV (TAR). Paralelamente a todos estos cambios hemos sido testigos de las épocas de esplendor de un buen número de ARV y de su posterior desaparición o de una disminución muy importante de su utilización debido, fundamentalmente, a sus efectos adversos y a la disponibilidad de nuevos fármacos con claras ventajas frente a aquellos (tolerabilidad, comodidad de administración, etc.). Todo ello ha permitido que la eficacia del tratamiento haya ido aumentando paulatinamente.
En el paciente que inicia por primera vez TAR (naïve), las posibilidades terapéuticas son amplias, pero el tratamiento está muy estandarizado y se basa en la abundante información científica disponible, que ha permitido elaborar unas sólidas recomendaciones. Todas las guías de tratamiento actuales de los países occidentales acerca de las pautas recomendadas en los pacientes naïve incluyen la combinación de 2 inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN) asociados a un tercer ARV de otra familia. Los inhibidores de la transcriptasa inversa no análogos de nucleósidos (ITINAN) juegan un papel fundamental en el tratamiento inicial. A pesar de las múltiples posibilidades terapéuticas disponibles en nuestro medio, en la práctica clínica habitual alrededor del 60–70% de los pacientes que inician tratamiento toman una pauta basada en ITINAN, fundamentalmente la combinación de tenofovir (TDF), emtricitabina (FTC) y efavirenz (EFV). Esta combinación, además de su elevada potencia y eficacia clínica, ha permitido, por primera vez, la administración de todo el TAR en un solo comprimido al día, lo cual ha supuesto un importante hito en el tratamiento de la infección por el VIH, facilitando la adaptación del TAR al estilo de vida del paciente y la adherencia terapéutica. En muchos de los pacientes que no inician TAR con esta pauta, el motivo es porque no se considera adecuado administrar EFV por razones tales como las resistencias primarias, las alteraciones neuropsiquiátricas asociadas, la baja barrera genética, las interacciones farmacocinéticas y el embarazo o deseo gestacional.
Rilpivirina (RPV) es un nuevo ITINAN de segunda generación, recientemente incorporado al arsenal terapéutico, muy atractivo para el tratamiento de los pacientes con infección por VIH, dado que por sus características resuelve muchos de los problemas planteados con los ITINAN de primera generación, además de su excelente comodidad de administración, tanto en su presentación individual (Edurant®) como asociado a TDF/FTC en un único comprimido a dosis fijas (Eviplera®).
En el presente capítulo se analiza la información clínica de RPV en pacientes naïve, que se ha evaluado fundamentalmente en 1 estudio fase IIb (TMC278-C204) y en 3 estudios fase III (ECHO [Early Capture HIV-1-infected adults], THRIVE [Transcriptase Inhibitors in Treatment-naïve Adults Infected with HIV-1] y STaR [Single-Tablet Regimen]).
Ensayo clínico fase IIbEl Estudio TMC278-C2041 es un ensayo clínico fase IIb, aleatorizado, a 96 semanas, en el que se comparó la eficacia de 3 dosis de RPV (25, 75 y 150mg) frente a EFV, asociados a 2 ITIAN definidos por el investigador (zidovudina/lamivudina [AZT/3TC] o TDF/FTC) en pacientes naïve. La aleatorización se estratificó en función de la región de origen y de los ITIAN acompañantes. Las 3 dosis de RPV se administraron a doble ciego, mientras que el grupo de EFV fue abierto.
Iniciaron tratamiento un total de 368 pacientes (93 RPV 25mg, 85 RPV 75mg, 91 RPV 150mg y 89 EFV). La proporción de pacientes con carga viral (CV) ≤ 50 copias/ml a las 48 semanas de tratamiento (objetivo primario) fue similar entre los 3 grupos de RPV (79,6, 80 y 76,9%) y comparable a la de EFV (80,9%). A las 96 semanas, la eficacia continuó siendo similar en los 4 grupos (76, 72, 71 y 73%, respectivamente), y también la tasa de fallo virológico (8,6, 9,5, 6,6 y 7,9%, respectivamente). No se observaron diferencias significativas ni a las 48 ni a las 96 semanas en la eficacia terapéutica ni en la proporción de pacientes con fallo virológico. Inicialmente se eligió la dosis de RPV de 75mg/día para continuar el desarrollo clínico.
En una primera fase de extensión del estudio TMC278-C204 (semanas 96 a 144), los pacientes de los 3 grupos de RPV pasaron a recibir 75mg/día. A las 144 semanas de tratamiento el 65% (180/279) de los paciente tratados con RPV y el 62% (55/89) del grupo de EFV presentaron CV ≤ 50 copias/ml (ITT-TLOVR).
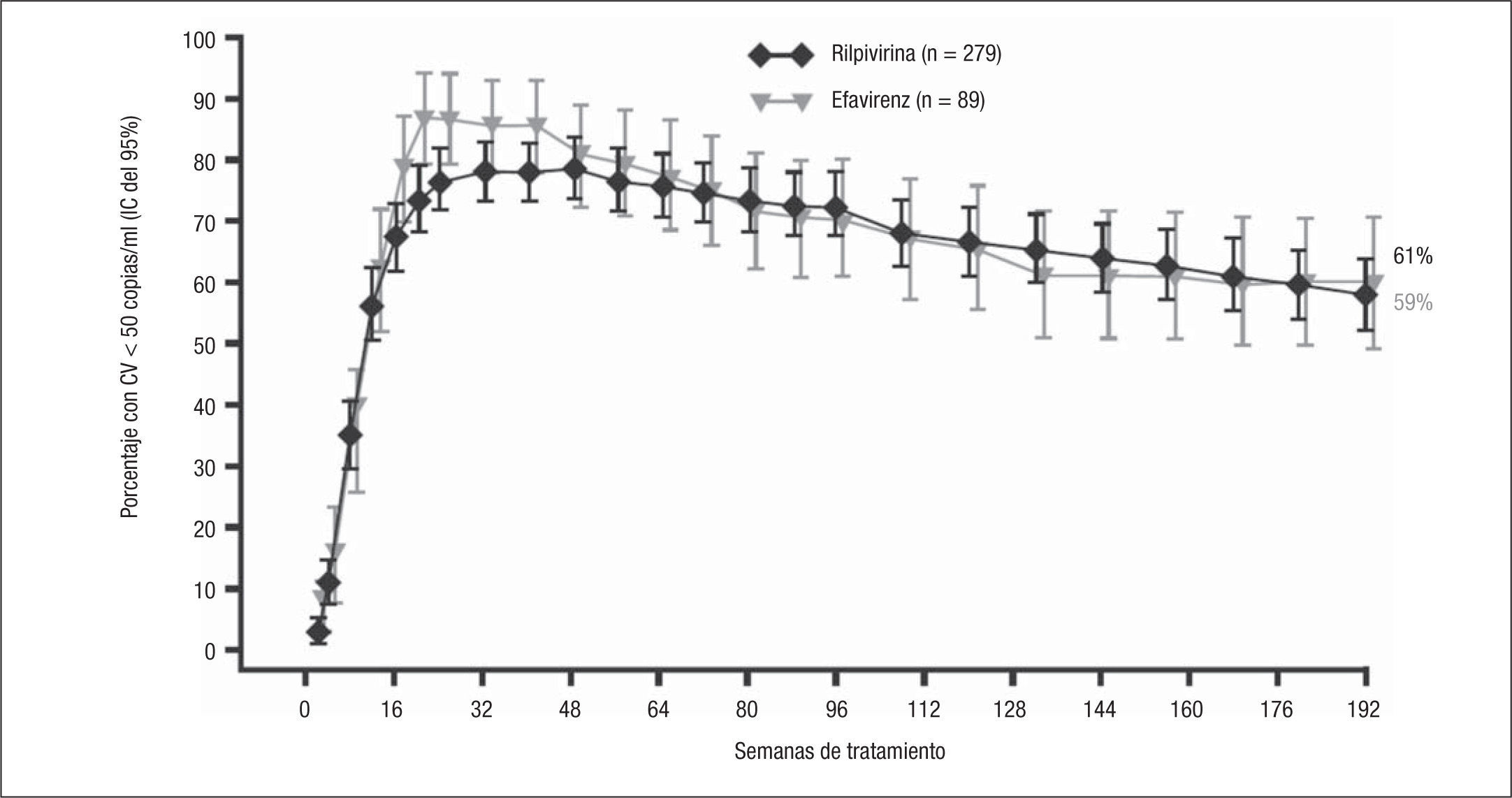
En los pacientes del estudio de fase IIb se observó una prolongación del intervalo QT corregido en el electrocardiograma con dosis de RPV de 75mg/día y de 300mg/día, que no apareció con 25mg/día, por lo cual, ante la igualdad de eficacia, la dosis de RPV que finalmente se eligió fue la de 25mg/día. Esta es la dosis que se administró a todos los pacientes en una segunda fase de extensión del estudio TMC278-C204 (semanas 144 a 240). A las 192 semanas de tratamiento, el 59% (164/279) de los pacientes tratados con RPV y el 61% (54/89) de los tratados con EFV continuaron con CV < 50 copias/ml (fig. 1)2. El porcentaje de pacientes con fallo virológico fue reducido (el 12 frente al 8%) y la mayoría de ellos ocurrieron en las fases iniciales del tratamiento.
Ensayos clínicos fase IIIEstudios ECHO y THRIVE 3,4 < 50 copias/ml hasta las 192 semanas (ITT TLOVR). IC: intervalo de confianza.")
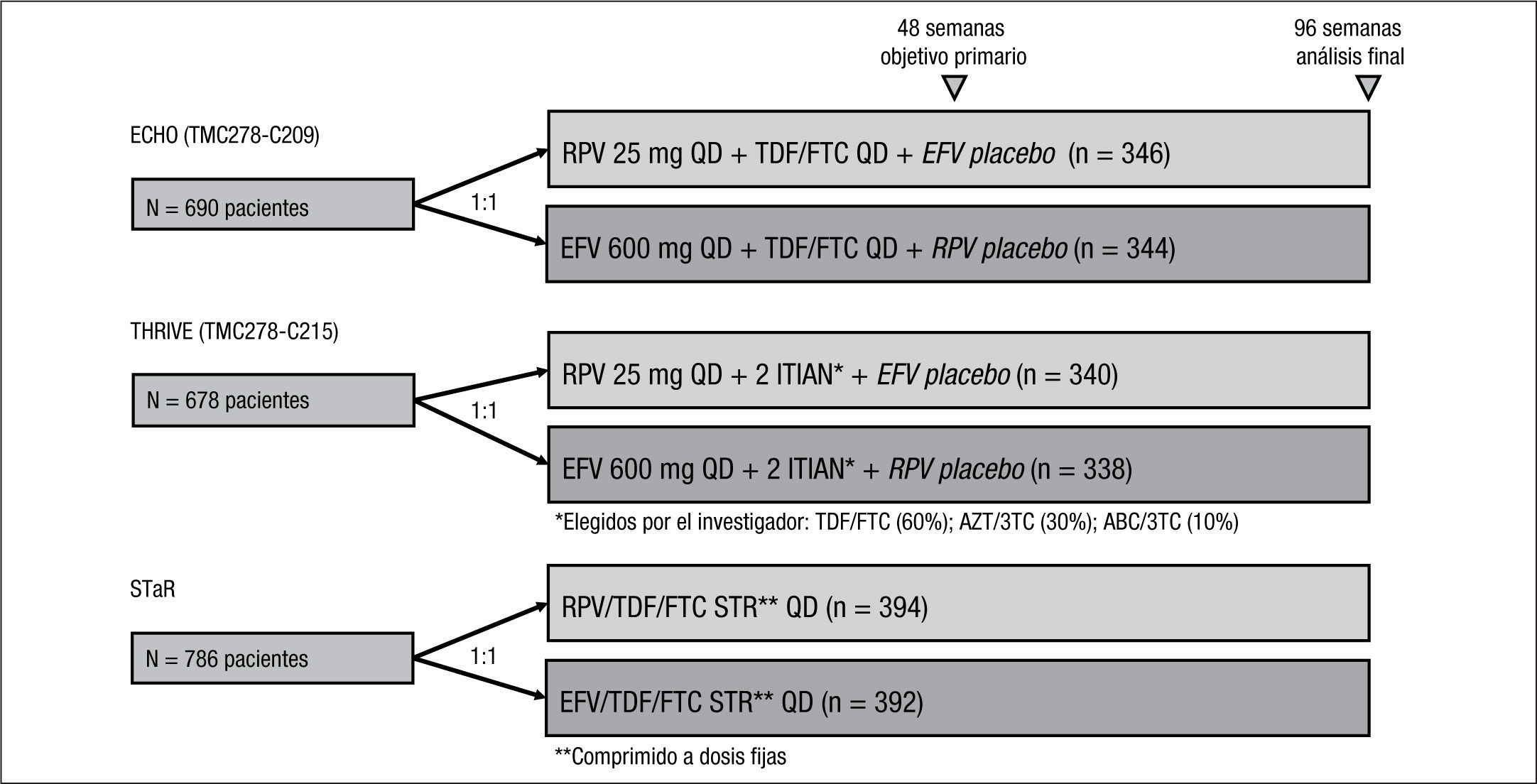
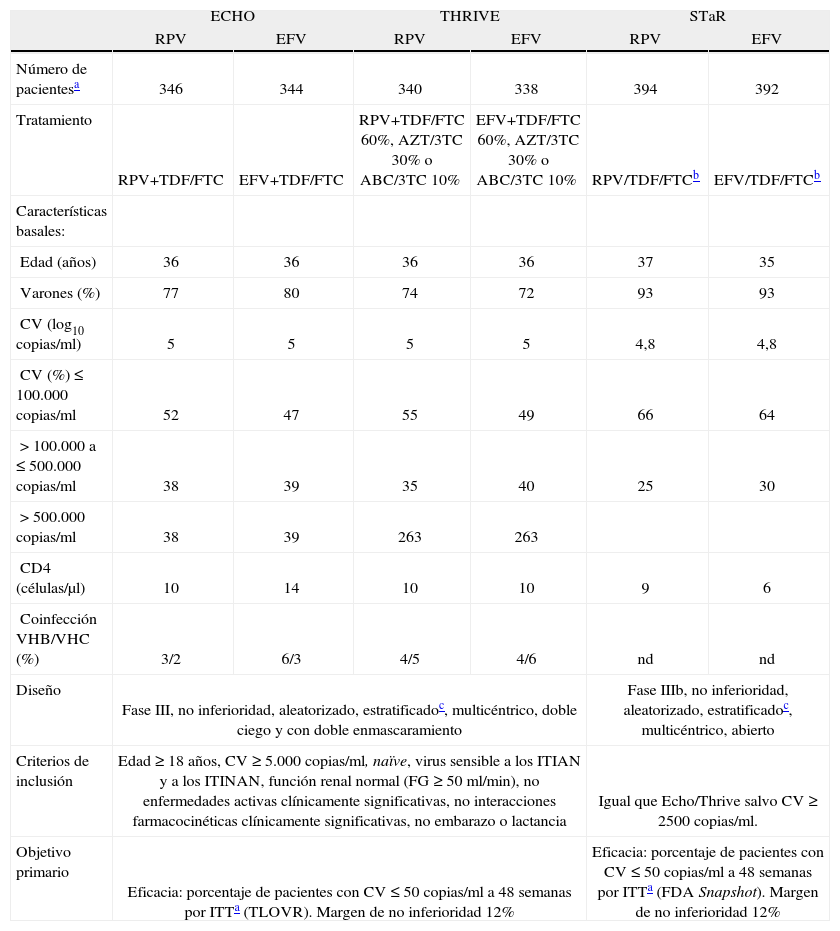
Son 2 estudios fase III, de no inferioridad, aleatorizados, estratificados según CV basal (≤ 100.000 copias/ml, de 100.000 a 500.000 copias/ml y > 500.000 copias/ml), multicéntricos, doble ciego y con doble enmascaramiento en pacientes sin tratamiento previo, con un diseño y unos criterios de inclusión y exclusión casi idénticos (fig. 2; tabla 1).
 con efavirenz (EFV) en pacientes con infección por VIH que no habían recibido tratamiento previo.")
Características basales y diseño de los ensayos clínicos fase III en los que se compara rilpivirina (RPV) con efavirenz (EFV) en pacientes con infección por virus de la inmunodeficiencia humana que no habían recibido tratamiento previo.
| ECHO | THRIVE | STaR | ||||
| RPV | EFV | RPV | EFV | RPV | EFV | |
| Número de pacientesa | 346 | 344 | 340 | 338 | 394 | 392 |
| Tratamiento | RPV+TDF/FTC | EFV+TDF/FTC | RPV+TDF/FTC 60%, AZT/3TC 30% o ABC/3TC 10% | EFV+TDF/FTC 60%, AZT/3TC 30% o ABC/3TC 10% | RPV/TDF/FTCb | EFV/TDF/FTCb |
| Características basales: | ||||||
| Edad (años) | 36 | 36 | 36 | 36 | 37 | 35 |
| Varones (%) | 77 | 80 | 74 | 72 | 93 | 93 |
| CV (log10 copias/ml) | 5 | 5 | 5 | 5 | 4,8 | 4,8 |
| CV (%) ≤ 100.000 copias/ml | 52 | 47 | 55 | 49 | 66 | 64 |
| > 100.000 a ≤ 500.000 copias/ml | 38 | 39 | 35 | 40 | 25 | 30 |
| > 500.000 copias/ml | 38 | 39 | 263 | 263 | ||
| CD4 (células/μl) | 10 | 14 | 10 | 10 | 9 | 6 |
| Coinfección VHB/VHC (%) | 3/2 | 6/3 | 4/5 | 4/6 | nd | nd |
| Diseño | Fase III, no inferioridad, aleatorizado, estratificadoc, multicéntrico, doble ciego y con doble enmascaramiento | Fase IIIb, no inferioridad, aleatorizado, estratificadoc, multicéntrico, abierto | ||||
| Criterios de inclusión | Edad ≥ 18 años, CV ≥ 5.000 copias/ml, naïve, virus sensible a los ITIAN y a los ITINAN, función renal normal (FG ≥ 50 ml/min), no enfermedades activas clínicamente significativas, no interacciones farmacocinéticas clínicamente significativas, no embarazo o lactancia | Igual que Echo/Thrive salvo CV ≥ 2500 copias/ml. | ||||
| Objetivo primario | Eficacia: porcentaje de pacientes con CV ≤ 50 copias/ml a 48 semanas por ITTa (TLOVR). Margen de no inferioridad 12% | Eficacia: porcentaje de pacientes con CV ≤ 50 copias/ml a 48 semanas por ITTa (FDA Snapshot). Margen de no inferioridad 12% | ||||
ABC/3TC: abacabir más lamivudina; CV: carga viral; FG: filtrado glomerular estimado; ITIAN: inhibidores de la transcriptasa inversa análogos de nucleós(t)idos; ITINAN: inhibidores de la transcriptasa inversa no análogos de nucleósidos; ITT: intención de tratamiento; nd: no disponible; TDF/FTC: tenofovir más emtricitabina; AZT/3TC: zidovudina más lamivudina.
Los resultados se expresan como medias, a menos que se indique porcentaje o número de casos.
RPV se administró con alimentos y EFV en ayunas. En ellos se comparó la eficacia y la seguridad de RPV (25mg 1 vez al día) frente a EFV (600mg 1 vez al día), ambos en combinación con 2 ITIAN. La principal diferencia entre ambos estudios fue la combinación de ITIAN. En el estudio ECHO todos los pacientes recibieron TDF y FTC a dosis fija (Truvada®), mientras que en el estudio THRIVE el investigador eligió los ITIAN entre 3 combinaciones posibles: TDF/FTC (60%), AZT/3TC (30%) o abacavir (ABC)/3TC (10%). En este estudio la aleatorización también se estratificó en función de los ITIAN administrados.
El objetivo primario de ambos estudios fue el porcentaje de pacientes que recibió, al menos, 1 dosis del fármaco en estudio, que alcanzó una CV ≤ 50 copias/ml a las 48 semanas. El análisis se realizó por intención de tratamiento (ITT) mediante el algoritmo TLOVR, considerándose no respondedores a los pacientes que interrumpieron el tratamiento por cualquier motivo o que presentaron fallo virológico (CV > 50 copias/ml confirmada). El margen de no inferioridad se estableció en el 12% (límite inferior del intervalo de confianza [IC] del 95%). Además se realizaron varios análisis diferentes: a) por protocolo, excluyendo las violaciones de protocolo (ITT-PP); b) ajustado por otros factores (ITT-modelo); c) excluyendo las interrupciones de tratamiento por razones distintas del fallo virológico (ITT-EV); d) según adherencia > o ≤ 95% (cuestionario M-MSRI), y e) ITT-FDA snapshot. Teniendo en cuenta la similitud de diseño se planificó un análisis conjunto de los 2 estudios a las 48 y a las 96 semanas5,6.
Las características basales fueron similares en ambos estudios y en ambos grupos de tratamiento (tabla 1). Se incluyeron e iniciaron tratamiento un total de 690 pacientes en el estudio ECHO y de 678 en el estudio THRIVE, con una edad media global de 36 años, un 25% de mujeres, una CV basal media de 5 logaritmos y una cifra basal media de CD4 de 254 por μl.
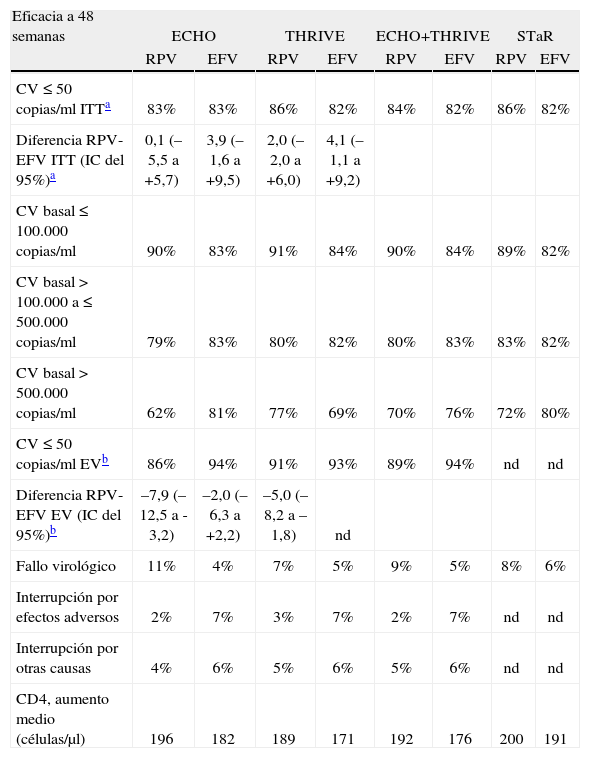
Datos de eficacia. La eficacia terapéutica a las 48 semanas (objetivo primario) resultó muy elevada en ambos grupos (tabla 2). RPV demostró no inferioridad frente a EFV, tanto en el estudio ECHO (el 83% con ambos tratamientos; IC del 95%, –5,5 a +5,7), como en el estudio THRIVE (el 86 frente al 82%; diferencia 3,9%; IC del 95%, –1,6 a +9,5) y en el análisis conjunto (el 84 frente al 82%; diferencia 2,0%; IC del 95%, –2,0 a +6,0). En los análisis por protocolo y ajustados por otros factores, los resultados fueron superponibles a los del objetivo primario.
Ensayos clínicos de fase III en pacientes sin tratamiento previo: eficacia terapéutica del tratamiento con rilpivirina (RPV) comparado con efavirenz (EFV), asociados a 2 inhibidores de la transcriptasa inversa análogos de nucleós(t)idos (ITIAN), a las 48 semanas.
| Eficacia a 48 semanas | ECHO | THRIVE | ECHO+THRIVE | STaR | ||||
| RPV | EFV | RPV | EFV | RPV | EFV | RPV | EFV | |
| CV ≤ 50 copias/ml ITTa | 83% | 83% | 86% | 82% | 84% | 82% | 86% | 82% |
| Diferencia RPV-EFV ITT (IC del 95%)a | 0,1 (–5,5 a +5,7) | 3,9 (–1,6 a +9,5) | 2,0 (–2,0 a +6,0) | 4,1 (–1,1 a +9,2) | ||||
| CV basal ≤ 100.000 copias/ml | 90% | 83% | 91% | 84% | 90% | 84% | 89% | 82% |
| CV basal > 100.000 a ≤ 500.000 copias/ml | 79% | 83% | 80% | 82% | 80% | 83% | 83% | 82% |
| CV basal > 500.000 copias/ml | 62% | 81% | 77% | 69% | 70% | 76% | 72% | 80% |
| CV ≤ 50 copias/ml EVb | 86% | 94% | 91% | 93% | 89% | 94% | nd | nd |
| Diferencia RPV-EFV EV (IC del 95%)b | –7,9 (–12,5 a -3,2) | –2,0 (–6,3 a +2,2) | –5,0 (–8,2 a –1,8) | nd | ||||
| Fallo virológico | 11% | 4% | 7% | 5% | 9% | 5% | 8% | 6% |
| Interrupción por efectos adversos | 2% | 7% | 3% | 7% | 2% | 7% | nd | nd |
| Interrupción por otras causas | 4% | 6% | 5% | 6% | 5% | 6% | nd | nd |
| CD4, aumento medio (células/μl) | 196 | 182 | 189 | 171 | 192 | 176 | 200 | 191 |
CV: carga viral de VIH; IC: intervalo de confianza; nd: no disponible.
Analizando las causas de fallo terapéutico a las 48 semanas, se observó que más pacientes tratados con EFV interrumpieron el tratamiento por efectos adversos (el 7% en la rama de EFV frente al 2% en la rama de RPV), mientras que más pacientes tratados con RPV presentaron fallo virológico (el 9% para RPV frente al 5% para EFV). En el análisis de eficacia, excluyendo a los pacientes que interrumpieron el tratamiento por causas diferentes al fallo virológico, RPV resultó menos eficaz que EFV en el estudio ECHO, pero siguió demostrando no inferioridad en el estudio THRIVE y en el análisis conjunto (tabla 2). Las diferencias en la proporción de pacientes con fallo virológico del estudio ECHO solo se observaron en pacientes con CV basal elevada. En el análisis conjunto de ambos estudios, en los pacientes con CV basal ≤ 100.000 copias/ml, la eficacia terapéutica de RPV resultó significativamente superior a la de EFV (el 90 frente al 84%; diferencia 6,6%; IC del 95%, 1,6–11,5), con la misma proporción de fallos virológicos (el 5% en cada grupo) y una mejor tolerabilidad de RPV7.
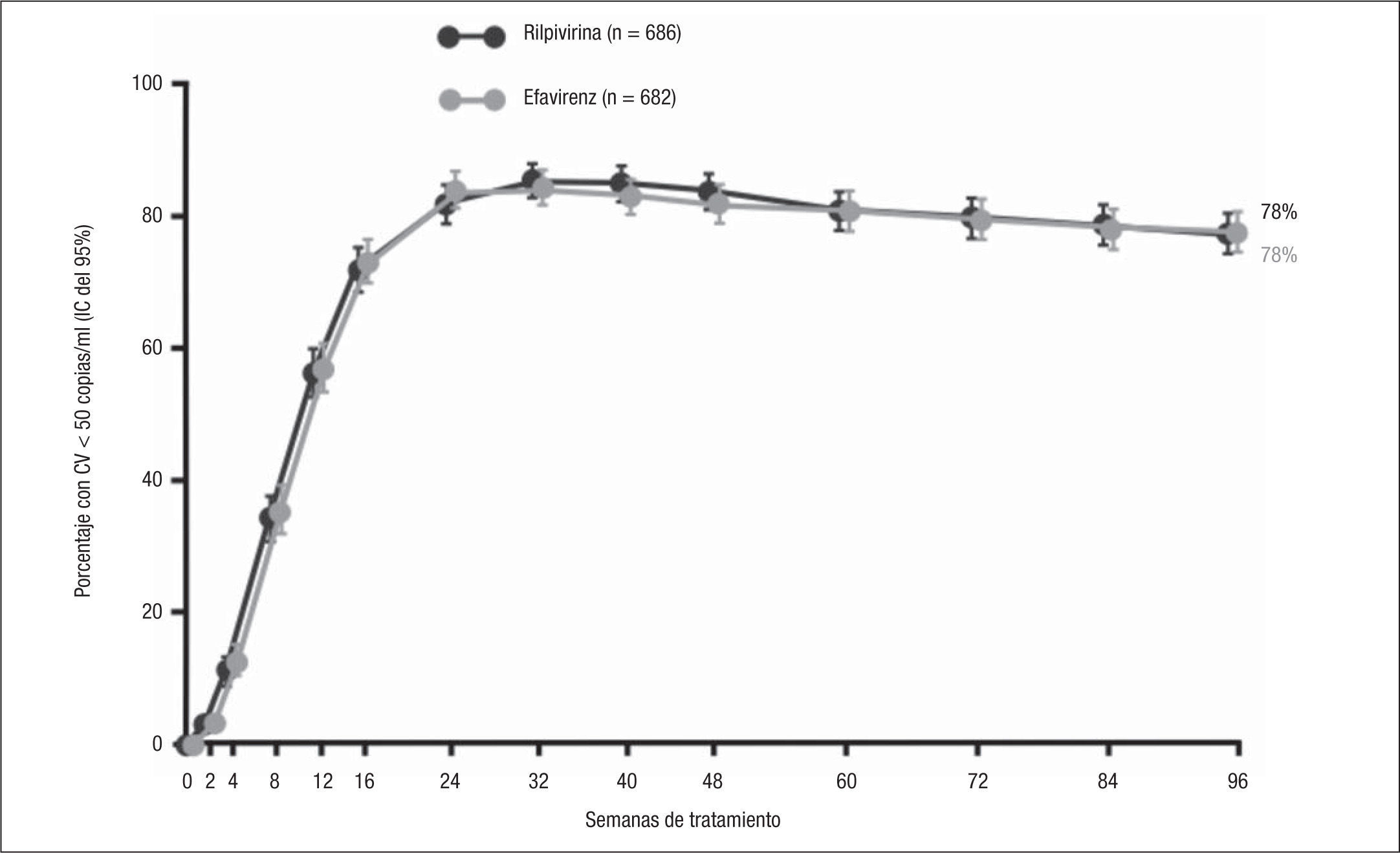
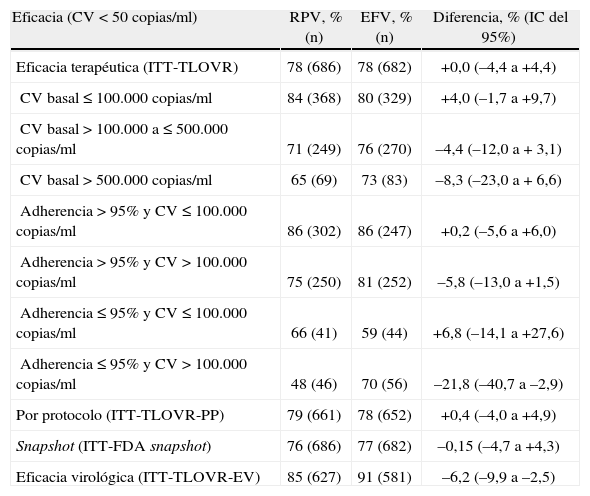
A las 96 semanas de tratamiento se mantuvo la elevada eficacia terapéutica (fig. 3). Globalmente, el 78% de los pacientes de cada grupo presentó una CV ≤ 50 copias/ml (ITT-TLOVR), demostrándose la no inferioridad de RPV en comparación con EFV (diferencia –0,0%; IC del 95%, –4,4 a +4,4) (tabla 3). La no inferioridad también se demostró en los estudios ECHO y THRIVE por separado. No se observaron diferencias significativas en la eficacia de RPV comparada con la de EFV en función de los ITIAN acompañantes, del sexo, de la edad, ni de la raza8. El número de pacientes coinfectados por virus de la hepatitis B (VHB) o C (VHC) en estos estudios es pequeño, observándose una eficacia terapéutica algo inferior que en pacientes no coinfectados, pero sin diferencias entre RPV y EFV9.
 < 50 copias/ml hasta las 96 semanas (ITT TLOVR). IC: intervalo de confianza.")
Eficacia de rilpivirina (RPV) comparada con efavirenz (EFV) a las 96 semanas de tratamiento. Análisis conjunto de los estudios ECHO y THRIVE.
| Eficacia (CV < 50 copias/ml) | RPV, % (n) | EFV, % (n) | Diferencia, % (IC del 95%) |
| Eficacia terapéutica (ITT-TLOVR) | 78 (686) | 78 (682) | +0,0 (–4,4 a +4,4) |
| CV basal ≤ 100.000 copias/ml | 84 (368) | 80 (329) | +4,0 (–1,7 a +9,7) |
| CV basal > 100.000 a ≤ 500.000 copias/ml | 71 (249) | 76 (270) | –4,4 (–12,0 a + 3,1) |
| CV basal > 500.000 copias/ml | 65 (69) | 73 (83) | –8,3 (–23,0 a + 6,6) |
| Adherencia > 95% y CV ≤ 100.000 copias/ml | 86 (302) | 86 (247) | +0,2 (–5,6 a +6,0) |
| Adherencia > 95% y CV > 100.000 copias/ml | 75 (250) | 81 (252) | –5,8 (–13,0 a +1,5) |
| Adherencia ≤ 95% y CV ≤ 100.000 copias/ml | 66 (41) | 59 (44) | +6,8 (–14,1 a +27,6) |
| Adherencia ≤ 95% y CV > 100.000 copias/ml | 48 (46) | 70 (56) | –21,8 (–40,7 a –2,9) |
| Por protocolo (ITT-TLOVR-PP) | 79 (661) | 78 (652) | +0,4 (–4,0 a +4,9) |
| Snapshot (ITT-FDA snapshot) | 76 (686) | 77 (682) | –0,15 (–4,7 a +4,3) |
| Eficacia virológica (ITT-TLOVR-EV) | 85 (627) | 91 (581) | –6,2 (–9,9 a –2,5) |
CV: carga viral; IC: intervalo de confianza; ITT: intención de tratamiento.
El análisis por protocolo y el análisis ITT-snapshot ofrecen resultados muy similares a los del análisis principal (tabla 3). Si se excluye a los pacientes que interrumpieron tratamiento por razones distintas al fallo virológico (ITT-EV), la eficacia virológica resultó inferior con RPV frente a EFV (el 85 frente al 91%; diferencia –6,2%; IC del 95%, –9,9 a –2,5). La adherencia subóptima y la CV basal elevada fueron los factores pronósticos más importantes, asociándose a una menor eficacia y a una mayor tasa de fallos virológicos en ambos grupos, resultando las diferencias más importantes con RPV que con EFV (tabla 3). La inmunodepresión profunda también se asoció a una menor eficacia, especialmente en el grupo de RPV, pero su importancia fue menor que la de la CV basal.
El incremento medio de linfocitos CD4 (IC del 95%) a las 96 semanas fue de 228 (215 a 240) células/μl con RPV y de 219 (206 a 233) células/μl con EFV.
Datos de seguridad.- -
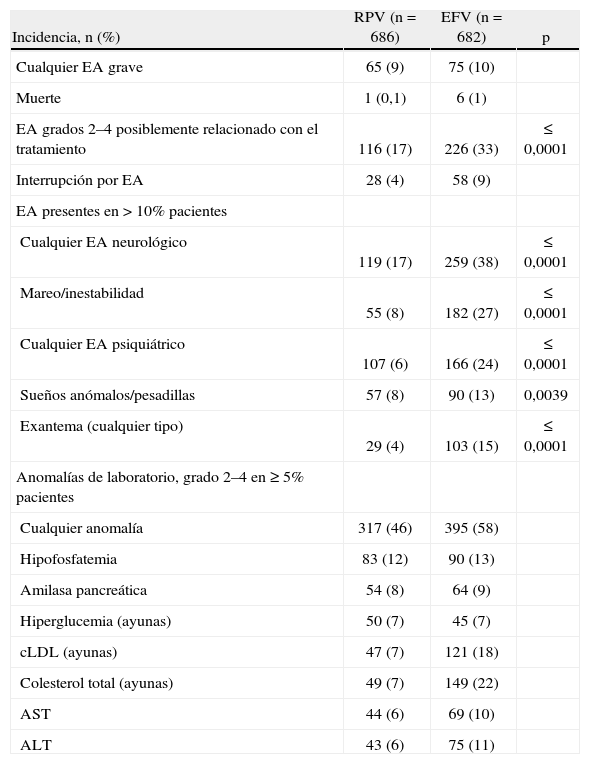
Tolerabilidad. El perfil de seguridad y tolerabilidad de RPV, a la dosis de 25mg utilizada en los ensayos clínicos, fue muy bueno. Las tasas de efectos adversos de grados 2–4 y las interrupciones por toxicidad fueron bajas (tabla 2). En los estudios ECHO y THRIVE, los efectos adversos de grados 2–4 con una relación, al menos posible con el tratamiento, fueron significativamente menores en la rama de RPV que en la rama de EFV (el 16 frente al 31%; p≤0,0001)3,4. Asimismo, la tasa de interrupciones por toxicidad fue también inferior con RPV que con EFV (el 3 frente al 8%). Los efectos adversos de grados 2–4, que tenían una relación al menos posible con el tratamiento, de mayor frecuencia (observados en > 2% de los pacientes de cualquier rama) fueron exantema, inestabilidad, sueños anómalos y pesadillas, cefalea, insomnio y náuseas (tabla 4)3,4. Todos ellos fueron menos frecuentes en los pacientes que recibían RPV. Los efectos adversos relacionados con el tratamiento que afectaron a ≥ 10% de los pacientes en cada grupo fueron la inestabilidad, los sueños anómalos y/o pesadillas, y el exantema. Todos ellos fueron significativamente menos frecuentes en pacientes expuestos a RPV que en los expuestos a EFV. La máxima incidencia de exantema se observó en las primeras 4 semanas de tratamiento y fue también significativamente menor en el grupo de RPV (tabla 4). La mayoría de los exantemas eran moderados (grados 1 o 2) y no hubo ningún exantema de grado 45.
Tabla 4.Efectos adversos (EA) y anomalías de laboratorio en los estudios ECHO y THRIVE.
Incidencia, n (%) RPV (n=686) EFV (n=682) p Cualquier EA grave 65 (9) 75 (10) Muerte 1 (0,1) 6 (1) EA grados 2–4 posiblemente relacionado con el tratamiento 116 (17) 226 (33) ≤ 0,0001 Interrupción por EA 28 (4) 58 (9) EA presentes en > 10% pacientes Cualquier EA neurológico 119 (17) 259 (38) ≤ 0,0001 Mareo/inestabilidad 55 (8) 182 (27) ≤ 0,0001 Cualquier EA psiquiátrico 107 (6) 166 (24) ≤ 0,0001 Sueños anómalos/pesadillas 57 (8) 90 (13) 0,0039 Exantema (cualquier tipo) 29 (4) 103 (15) ≤ 0,0001 Anomalías de laboratorio, grado 2–4 en ≥ 5% pacientes Cualquier anomalía 317 (46) 395 (58) Hipofosfatemia 83 (12) 90 (13) Amilasa pancreática 54 (8) 64 (9) Hiperglucemia (ayunas) 50 (7) 45 (7) cLDL (ayunas) 47 (7) 121 (18) Colesterol total (ayunas) 49 (7) 149 (22) AST 44 (6) 69 (10) ALT 43 (6) 75 (11) ALT: alanino aminotransferasa; AST: aspartato aminotransferasa; cLDL: colesterol unido a lipoproteínas de baja densidad; EFV: efavirenz; RPV: rilpivirina.
- -
Perfil metabólico y de laboratorio. Desde el punto de vista de toxicidad de laboratorio se observó una incidencia significativamente inferior de elevación de aspartato aminotransferasa (AST) grados 2–4 y de alanina transferasa (ALT) en pacientes expuestos a RPV, en comparación con los expuestos a EFV (tabla 4). Por el contrario, la incidencia de elevación de grados 2–3 de bilirrubina total fue significativamente mayor en los pacientes que recibían RPV. No obstante, la incidencia de efectos adversos hepáticos graves fue baja en ambas ramas de tratamiento y, entre ellas, menor en la rama de RPV. De hecho, no se observó ningún efecto adverso hepático grave relacionado con el tratamiento que precisara su interrupción en la rama de RPV, mientras que se observaron 2 casos en la rama de EFV. Como suele ser norma, la incidencia de efectos adversos hepáticos en ambas ramas de tratamiento fue mayor en pacientes con coinfección por VHB o VHC que en los pacientes sin coinfección
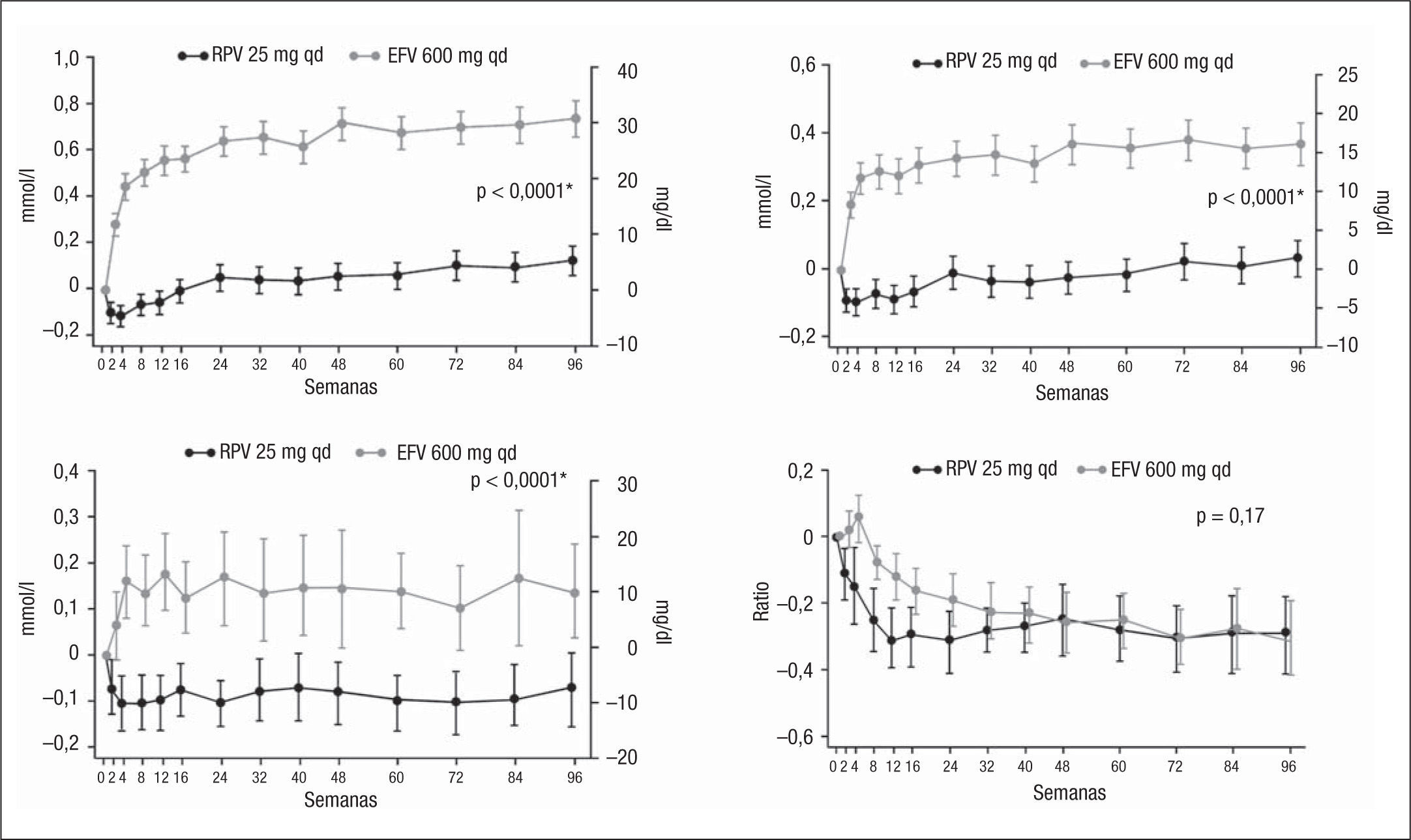
La exposición a RPV se asoció con menores incrementos de las concentraciones de colesterol total, colesterol unido a lipoproteínas de baja densidad (cLDL), colesterol unido a lipoproteínas de alta densidad (cHDL) y de triglicéridos que la exposición a EFV. De hecho, los valores medios de cLDL y de triglicéridos no aumentaron con respecto a las mediciones basales en los pacientes expuestos a RPV, mientras que en los expuestos a EFV se objetivó un incremento significativo de todos ellos (fig. 4)5. A pesar de dichos efectos, no se observaron cambios significativos en la ratio aterogénica colesterol total/cHDL a la semana 48, que era similar en ambos grupos.
![Cambio medio (intervalo de confianza [IC] del 95%) de las concentraciones de lípidos en ayunas desde el momento basal hasta la semana 96 en los estudios ECHO+THRIVE. A) Colesterol total. B) Colesterol unido a lipoproteínas de baja densidad (cLDL). C) Triglicéridos. D) Cociente colesterol total/colesterol unido a lipoproteínas de alta densidad (cHDL). EFV: efavirenz; RPV: rilpivirina; *Valor de p a la semana 96 (test de Wilcoxon).](https://static.elsevier.es/multimedia/0213005X/00000031000000S2/v1_201311170010/S0213005X13701393/v1_201311170010/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNc+34VOwP+2HTB5hao7CSfc3vHAcopEDD4TvJ2qcebX0k9HqN8G7zZXrpLvzzF4z81eceuP1VnErA/y1KGYPge25KaNW5DULV37ohwuvr416kEbcxpTr5UMTNCGpouAupX4EeDzlz7tWr0wMK6xwV6OXXFTQx+vLcDTwe/GMgvfqcchzqpOo9Af/ZVZ6subL7Ul5dVDebnxlncw262kPa02RuYWpg1O+DEjfwl3ijsv+9deLNprrtab0QnYuh4wDpIWWf4QixU2z1f3RHfiJ50m "Cambio medio (intervalo de confianza [IC] del 95%) de las concentraciones de lípidos en ayunas desde el momento basal hasta la semana 96 en los estudios ECHO+THRIVE. A) Colesterol total. B) Colesterol unido a lipoproteínas de baja densidad (cLDL). C) Triglicéridos. D) Cociente colesterol total/colesterol unido a lipoproteínas de alta densidad (cHDL). EFV: efavirenz; RPV: rilpivirina; *Valor de p a la semana 96 (test de Wilcoxon).")
Cambio medio (intervalo de confianza [IC] del 95%) de las concentraciones de lípidos en ayunas desde el momento basal hasta la semana 96 en los estudios ECHO+THRIVE. A) Colesterol total. B) Colesterol unido a lipoproteínas de baja densidad (cLDL). C) Triglicéridos. D) Cociente colesterol total/colesterol unido a lipoproteínas de alta densidad (cHDL). EFV: efavirenz; RPV: rilpivirina; *Valor de p a la semana 96 (test de Wilcoxon).
En los pacientes expuestos a RPV se observó un discreto incremento de las cifras de creatinina desde su valor basal, que no se observó en los pacientes expuestos a EFV5. Este incremento de creatinina en la rama de RPV se ha relacionado con cambios en la secreción tubular de creatinina sin efecto directo alguno sobre el filtrado glomerular. Ello se ha comprobado mediante la estimación del filtrado glomerular con otros métodos como la cistatina C. Sin embargo, no se documentó ningún caso de elevación grados 3–4 de creatinina asociado al uso de RPV. Estos cambios no se consideran clínicamente relevantes dado que no reflejan un cambio en la tasa de filtración glomerular y ninguno de los pacientes interrumpió el tratamiento debido a un aumento de creatinina sérica. Los aumentos de creatinina fueron similares con otros ITIAN3.
- -
Otras toxicidades. El uso de dosis supraterapéuticas de RPV (p. ej., 75, 150 o 300mg/día) se asoció en ensayos clínicos de fases I y II con prolongación del intervalo QT corregido1,10,11. No obstante, con la dosis finalmente aprobada de 25mg no se han observado cambios clínicamente significativos en la longitud del intervalo QT corregido, tanto cuando RPV se administró sola en estudios de fase I o combinada con ITIAN en los ensayos clínicos de fase III ECHO y THRIVE5,12.
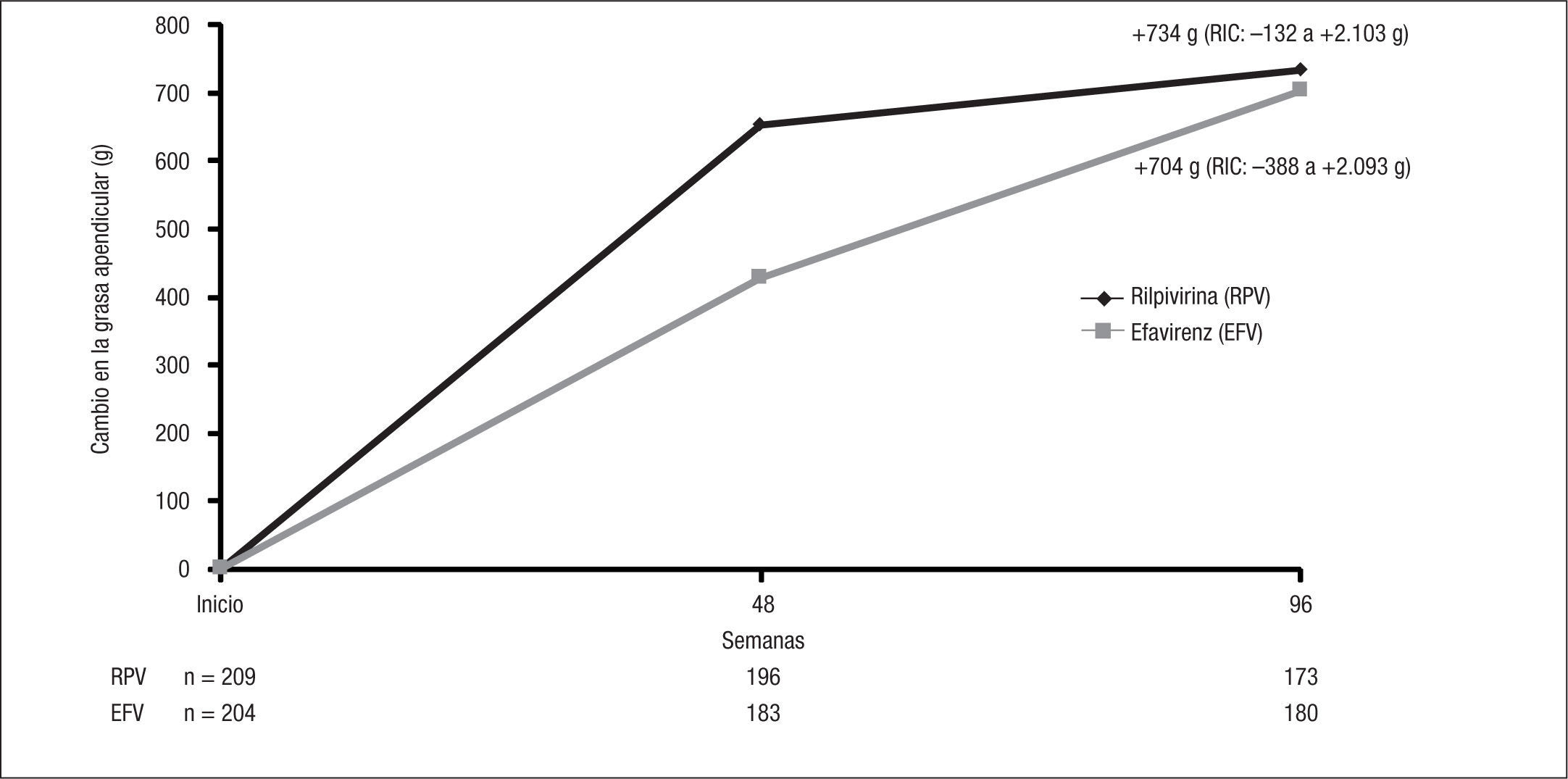
La existencia de datos in vitro en los que RPV no demostró efectos deletéreos sobre los adipocitos humanos con valores de fármaco similares a los conseguidos con las dosis de uso clínico hacía prever que dicho fármaco no exhibiría toxicidad sobre el tejido graso subcutáneo13. El subestudio metabólico de los estudios ECHO y THRIVE puso de manifiesto un incremento significativo de grasa subcutánea desde el momento basal hasta la semana 96 en ambas ramas del estudio, sin diferencias entre ellas (fig. 5)14.

Los efectos de RPV y EFV sobre la mineralización ósea se examinaron en un subestudio de los estudios ECHO y THRIVE, no demostrándose efectos deletéreos sobre la misma de ambos fármacos, más allá de los efectos que sobre la masa ósea comporta el inicio del TAR y la inclusión en dichas combinaciones de TDF14. Ello se observó a pesar del conocido impacto negativo de la administración de EFV sobre el metabolismo de la vitamina D y sobre sus niveles plasmáticos, extremo que se objetivó en un subestudio del estudio ECHO, en el que se observaron descensos significativamente más importantes en los pacientes randomizados a EFV que en los que habían sido randomizados a RPV15.
Datos de resistencias. Debido a las características intrínsecas a la molécula de RPV, a su mecanismo de unión con el bolsillo hidrofóbico de la transcriptasa inversa y a los datos experimentales obtenidos in vitro, se anticipó que RPV podía tener una barrera genética elevada in vivo16. No obstante, los análisis genotípicos de resistencia en pacientes en fracaso virológico procedentes de los estudios ECHO y THRIVE han demostrado que la selección de mutaciones de resistencia es frecuente en los pacientes que fracasan a un régimen de RPV más 2 ITIAN, especialmente en los pacientes que tenían una CV basal > 100.000 copias/ml17. Adicionalmente, la tasa de selección de resistencias a los ITIAN fue mayor en los pacientes que fracasaban con un régimen de RPV más 2 ITIAN que en los que fracasaban con un régimen de EFV más 2 ITIAN17. Globalmente, en pacientes que fracasaron y que tenían estudio genotípico disponible, 44 de 62 (71%) de los que fracasaron con RPV y 16 de 58 (57%) de los que fracasaron con EFV, presentaban nuevas mutaciones asociadas a resistencia. Entre los pacientes que fracasaron con RPV, el 38% de los pacientes con CV basal ≤ 100.000 copias/ml y el 72% de los que tenían CV basal > 100.000 copias/ml presentaron nuevas mutaciones asociadas a resistencia a los ITINAN, comparados con el 42 y el 63%, respectivamente, de los pacientes que fracasaron con EFV. Los porcentajes respectivos de resistencia a los ITIAN en pacientes con CV basal baja o elevada fueron del 44 y 76% para los expuestos a RPV más 2 ITIAN, en comparación con el 17 y 44% en pacientes que fracasaron con EFV más 2 ITIAN18,19. Por lo tanto, dentro de indicación (CV ≤ 100.000 copias/ml), el porcentaje de fracasos virológicos y de mutaciones de resistencia asociadas a ITINAN fue similar, aunque en la rama de RPV las mutaciones a ITIAN fueron ligeramente superiores.
Las mutaciones de resistencia a ITINAN más frecuentes que aparecieron en los fracasos a RPV fueron la E138K (45%), mutación muy infrecuente en los fracasos con otros ITINAN, seguida por la K101E (13%), H221Y (10%), V90I, Y181C y V189I (8% cada una de ellas). En lo que respecta a las mutaciones asociadas a resistencia a los ITIAN seleccionadas cuando hay fracaso con RPV más 2 ITIAN, la más frecuente fue la M184I (47%), seguida por la M184V (23%). La asociación entre E138K y M184I se observó en el 46% de los pacientes con fracaso a RPV17 y esta emergencia de mutantes dobles E138K/M184I parece relacionarse con que esta combinación confiere un elevado nivel de resistencia a RPV, comparado con la existencia de únicamente la E138K y también a una ventaja competitiva con respecto a la capacidad replicativa de los dobles mutantes comparada con la existencia aislada de la M184I o E138K/M184V18,19.
El perfil de mutaciones de RPV y etravirina (ETR) es similar y, por tanto, la existencia de resistencia cruzada entre ambos fármacos es esperable. Consecuentemente, los pacientes que fracasan a un régimen basado en RPV y desarrollan resistencia genotípica a RPV presentan resistencia cruzada con todos los ITINAN, incluyendo ETR (90%), EFV (87%) y nevirapina (48%), mientras que en pacientes que fracasan con EFV, solo se observa resistencia cruzada con respecto a nevirapina17.
Estudio STaR20Es un ensayo clínico fase IIIb, de no inferioridad, aleatorizado, estratificado según CV basal (≤ o > 100.000 copias/ml), multicéntrico, abierto, en pacientes sin tratamiento previo, en el que por primera vez se comparan 2 pautas de TAR administradas en un solo comprimido al día: RPV/TDF/FTC (n=394) frente a EFV/TDF/FTC (n=392). Las características basales fueron similares en ambos grupos (tabla 1). El objetivo primario fue el porcentaje de pacientes con CV≤50 copias/ml a las 48 semanas, por ITT mediante el análisis snapshot de la FDA, con un margen de no inferioridad del 12%.
Datos de eficacia. A las 48 semanas de tratamiento, el 86% de pacientes del grupo de RPV y el 82% del grupo de EFV presentaban una CV≤50 copias/ml (tabla 2) demostrándose la no inferioridad de RPV (diferencia +4,1%; IC del 95%, –1,1 a +9,2%). Presentaron CV < 50 copias/ml el 80 y el 82% de pacientes, respectivamente (diferencia –1,8%; IC del 95%, –11,1 a +7,5). En los pacientes con CV basal ≤100.000 copias/ml RPV/TDF/FTC resultó superior a EFV/TDF/FTC (CV≤50 copias/ml snapshot: el 89 y el 82%; diferencia +7,1%; IC del 95%, +1,1 a +13,4). Las tasas de fallo virológico resultaron bajas y similares en ambos grupos (el 8 frente al 6%), sin diferencias notables entre RPV y EFV en pacientes con CV basal ≤ 100.000 copias/ml (el 5 frente al 3%) ni con CV basal > 100.000 copias/ml (el 14 frente al 10%).
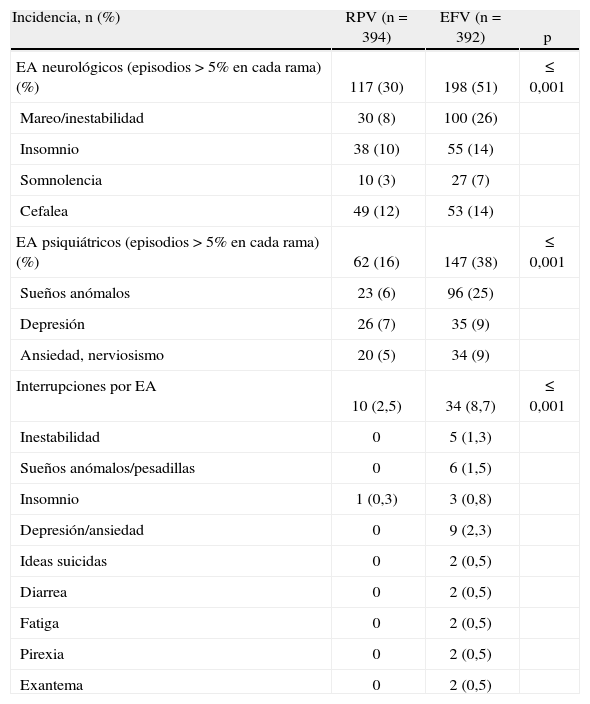
Datos de seguridad. El perfil de seguridad de las combinaciones utilizadas en el estudio STaR fue muy similar a los referidos para los estudios ECHO y THRIVE. Globalmente, la aparición de efectos adversos neuropsiquiátricos fue significativamente más frecuente en la rama de EFV que en la de RPV (tabla 5). La interrupción del tratamiento asignado hasta las 48 semanas de seguimiento ocurrió de forma significativamente más frecuente en la rama de EFV que en la de RPV (el 8,7 frente al 2,5%; p≤0,001). Dichas tasas de interrupción se debieron, en gran medida, a una mayor incidencia de efectos adversos neuropsiquiátricos entre los pacientes expuestos a EFV20. De hecho, los efectos adversos más frecuentes que motivaron la interrupción fueron la depresión o ansiedad (2,3%) seguida por los sueños anómalos o pesadillas (1,5%).
Efectos adversos (EA) en el estudio STaR.
| Incidencia, n (%) | RPV (n=394) | EFV (n=392) | p |
| EA neurológicos (episodios > 5% en cada rama) (%) | 117 (30) | 198 (51) | ≤ 0,001 |
| Mareo/inestabilidad | 30 (8) | 100 (26) | |
| Insomnio | 38 (10) | 55 (14) | |
| Somnolencia | 10 (3) | 27 (7) | |
| Cefalea | 49 (12) | 53 (14) | |
| EA psiquiátricos (episodios > 5% en cada rama) (%) | 62 (16) | 147 (38) | ≤ 0,001 |
| Sueños anómalos | 23 (6) | 96 (25) | |
| Depresión | 26 (7) | 35 (9) | |
| Ansiedad, nerviosismo | 20 (5) | 34 (9) | |
| Interrupciones por EA | 10 (2,5) | 34 (8,7) | ≤ 0,001 |
| Inestabilidad | 0 | 5 (1,3) | |
| Sueños anómalos/pesadillas | 0 | 6 (1,5) | |
| Insomnio | 1 (0,3) | 3 (0,8) | |
| Depresión/ansiedad | 0 | 9 (2,3) | |
| Ideas suicidas | 0 | 2 (0,5) | |
| Diarrea | 0 | 2 (0,5) | |
| Fatiga | 0 | 2 (0,5) | |
| Pirexia | 0 | 2 (0,5) | |
| Exantema | 0 | 2 (0,5) |
EFV: efavirenz; RPV: rilpivirina.
El perfil lipídico de los fármacos en estudio corroboró los datos de los estudios ECHO y THRIVE, observándose incrementos más importantes en colesterol total, cLDL, cHDL y triglicéridos en la rama de EFV con respecto a la de RPV. No obstante, el cambio de la ratio colesterol total/cHDL fue similar en ambas ramas de tratamiento20.
Datos de resistencias. El perfil de resistencias observado en el estudio STaR fue similar al de los pacientes en tratamiento con FTC/FTC junto con RPV o EFV de los estudios ECHO y THRIVE, con la diferencia que la emergencia global de resistencias fue menor en la rama de RPV (el 4 frente al 7%), básicamente por una menor frecuencia de la mutación E138K/Q (el 2 frente al 4%) y también se constató una menor aparición de resistencias entre los ITIAN (el 4 frente al 7%) en la rama de RPV20. Al igual que en los estudios ECHO y THRIVE, el porcentaje de aparición de resistencias durante el fracaso se incrementaba en la rama de RPV según el estrato de CV en el momento basal, siendo mayor cuanto mayor fuera esta.
Conclusiones- -
La dosis de uso de RPV en pacientes que inician tratamiento es de 25mg 1 vez al día, ya sea en un comprimido que solo contiene este fármaco (Edurant®) o en un comprimido que contiene RPV/TDF/FTC (Eviplera®).
- -
RPV ha demostrado una eficacia no inferior a la de EFV en ensayos clínicos de fase III en pacientes con infección por VIH sin experiencia terapéutica previa con ARV, ambos en combinación con 2 ITIAN.
- -
RPV ha mostrado una mejor tolerabilidad que EFV, especialmente en lo referente a efectos adversos del área neuropsiquiátrica, exantema cutáneo y de la esfera lipídica.
- -
En los pacientes con CV basal < 100.000 copias/ml, la eficacia terapéutica de RPV fue superior a la de EFV mostrando además un perfil más favorable de tolerabilidad, con una tasa de fallo virológico similar.
- -
Los datos del estudio ECHO no permitieron demostrar la no inferioridad con respecto a EFV en pacientes con CV basal > 100.000 copias/ml. La incidencia de fallo virológico en pacientes con CV basal elevada fue significativamente superior con RPV que con EFV en los pacientes del estudio ECHO, y en el análisis conjunto de los estudios ECHO y THRIVE. Según estos datos, la EMA y la FDA han aprobado el uso de RPV en pacientes con CV < 100.000 copias/ml.
- -
RPV ya se considera como integrante de pautas preferentes en algunas guías terapéuticas. Las guías DHHS21 (marzo de 2012) e IAS-USA22 (julio de 2012), en espera de datos adicionales, la consideran todavía como un régimen alternativo en pacientes naïve a ARV. Las guías europeas23 (noviembre de 2012) y las de GeSIDA (enero de 2013)24 la consideran como un régimen recomendado, limitándose a pacientes con CV basal < 100.000 copias/ml.
Este trabajo ha sido parcialmente realizado gracias a las siguientes ayudas: Fondo de Investigaciones Sanitarias (FIS PI08/00256, PI09/02123, PI10/2635 y PI12/01840), Ministerio de Sanidad, Política Social e Igualdad (EC11–293), Programa de Suport als Grups de Recerca AGAUR (2009 SGR 1061) y Red de Investigación en SIDA (RIS RD06/006/0022, RD06/0006/1004 y RD06/0006/0039, RD12/0017/0003). Pere Domingo recibe ayuda del Programa de Intensificación de Investigadores, Instituto de Salud Carlos III (INT12/383).
Conflicto de interesesLos autores han realizado asesorías o han recibido ayudas para investigación o docencia en relación con la infección por el VIH de los siguientes laboratorios farmacéuticos: Pere Domingo de Abbott, Boehringer Ingelheim, BMS, Gilead, GSK, Janssen-Cilag, MSD, Pfizer, ViiV, Recordati, Theratehcnologies y Ferrer International; Esteban Ribera de Abbott, Boehringer Ingelheim, BMS, Gilead, GSK, Janssen-Cilag, MSD, Pfizer, Roche Farma, Schering Plough y ViiV.

