


Hasta un 5% de los hiperparatiroidismos primarios (HPT) son hereditarios. La importancia de diagnosticar este tipo de HPT radica en que tienen presentaciones y asociaciones especiales y requieren, por tanto, aproximaciones especiales.
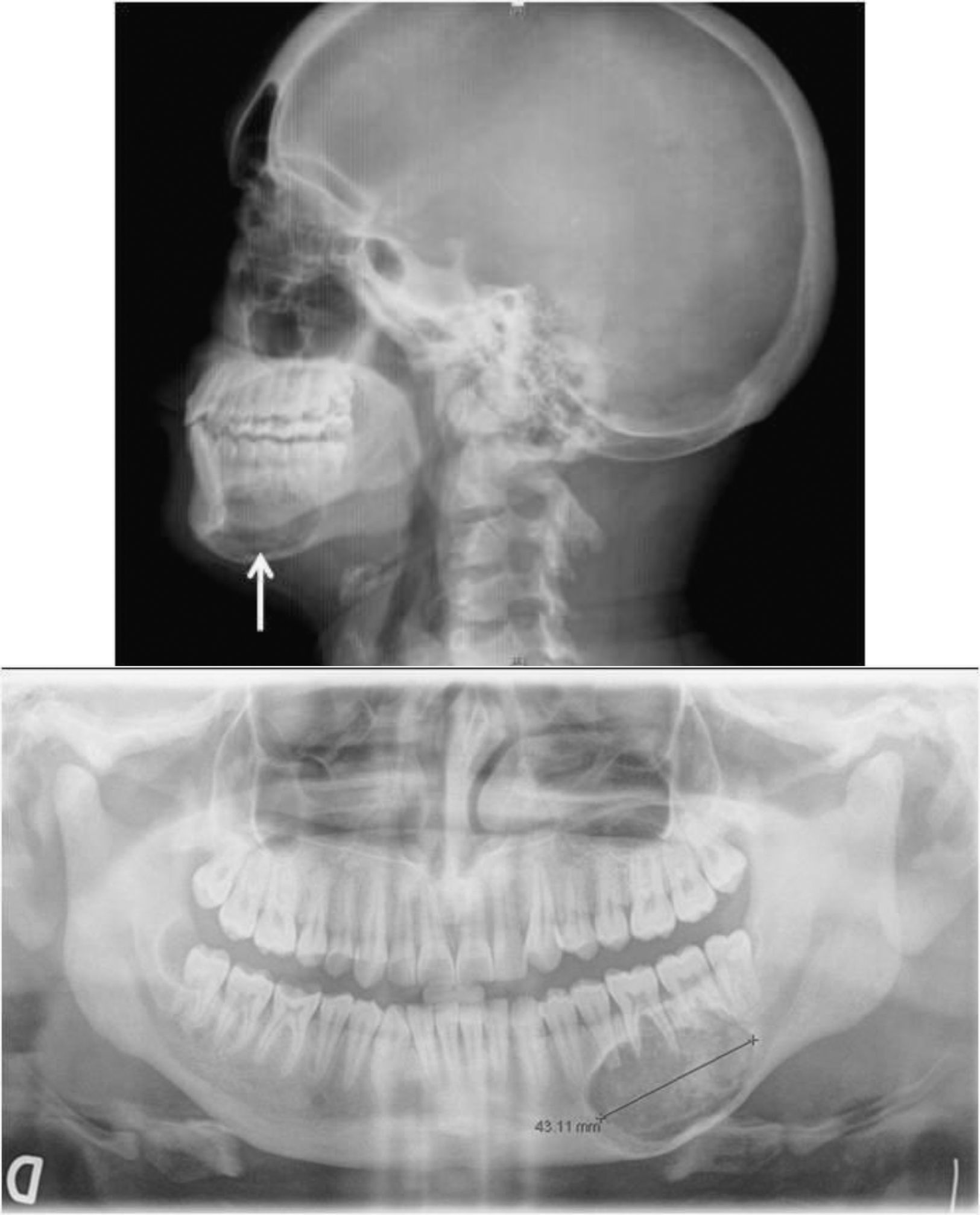
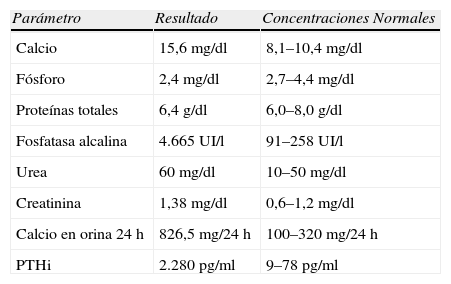
Varón de 27 años remitido por el hallazgo de calcio de 15,6mg/dl en estudio por astenia, anorexia, pérdida de peso y disminución de 5cm en la talla, acompañados en los últimos meses de dolor lumbar. Había sido estudiado por presentar una lesión mandibular indolora de meses de evolución, cuya biopsia era descrita como «lesión fibro-ósea del maxilar» (fig. 1). Entre los antecedentes familiares destacaba que una tía de su madre estaba operada de tiroides y las paratiroides, y que dos primos de su madre habían sido intervenidos de HPT (uno de ellos operado también de un tumor mandibular). Ni su madre ni ningún otro familiar de la línea materna estaban diagnosticados de HPT. Presentaba cifosis dorsal, lordosis lumbar y una tumoración dura de más de 3cm en la región laterocervical derecha, que se extendía retroesternalmente. En la tabla 1 se ven los hallazgos analíticos al ingreso. Se instauró tratamiento con sueroterapia y, posteriormente, diuréticos de asa, y 2 dosis de ácido zolendrónico (4mg i.v, diluidos en 10ml de suero fisiológico, pasados en 15min), manteniendo niveles de calcio alrededor de 12mg/dl. Se realizaron los siguientes estudios complementarios:
- 1.
Ecografía cervical: tumoración hipoecogénica polilobulada de 4,5×3×2,6cm laterocervical derecha. Adyacente al lóbulo tiroideo izquierdo, otra imagen nodular de 1cm de diámetro. Ambas lesiones eran altamente sugestivas de adenomas paratiroideos, sin poder descartarse la posibilidad de carcinoma.
- 2.
Tomografía computarizada cérvico toraco abdominal: signos de HPT avanzado con gran osteopenia, múltiples aplastamientos vertebrales y fracturas patológicas en las costillas. Litiasis renal bilateral, sin evidencia de masas renales.
- 3.
Serie ósea: lesión osteolítica en la mandíbula. Múltiples fracturas costales y del esternón, con engrosamiento de la pleura subyacente y esclerosis del esqueleto torácico. Esclerosis irregular de los bordes superior e inferior de los cuerpos vertebrales («vértebra en jersey de rugby») y acuñamiento de múltiples vértebras dorsales y lumbares. Osteopenia en sal y pimienta del esqueleto pélvico y femoral proximal.
- 4.
Densitometría ósea: Z score -2,46 en columna lumbar; Z score -3,59 en cuello femoral.
 y en la ortopantomografía (abajo). Además se aprecia ausencia de lámina dura de los dientes.")
Hallazgos analíticos al ingreso
| Parámetro | Resultado | Concentraciones Normales |
| Calcio | 15,6mg/dl | 8,1–10,4mg/dl |
| Fósforo | 2,4mg/dl | 2,7–4,4mg/dl |
| Proteínas totales | 6,4g/dl | 6,0–8,0g/dl |
| Fosfatasa alcalina | 4.665UI/l | 91–258UI/l |
| Urea | 60mg/dl | 10–50mg/dl |
| Creatinina | 1,38mg/dl | 0,6–1,2mg/dl |
| Calcio en orina 24h | 826,5mg/24h | 100–320mg/24h |
| PTHi | 2.280pg/ml | 9–78pg/ml |
PTHi: paratirina.
Fue sometido a tiroidectomía total más paratiroidectomía subtotal (paratiroides inferior derecha e inferior izquierda). Se confirmó la existencia de un carcinoma en la paratiroides (CP) inferior derecha de 5×4×3cm con infiltración de tejidos blandos adyacentes, sin evidencia de metástasis en ganglios regionales cervicales, así como un segundo CP en la glándula inferior izquierda, de 0,8cm. No se evidenció la presencia de patología tiroidea.
En el postoperatorio presentó hipocalcemia sintomática y prolongada de difícil control, en relación con el síndrome del hueso hambriento, a pesar del tratamiento con dosis altas de calcio y vitamina D. Transcurridos 18 meses desde la intervención el paciente tenía calcemias normales pero seguía precisando tratamiento con calcio y vitamina D, aunque las dosis se fueron reduciendo progresivamente.
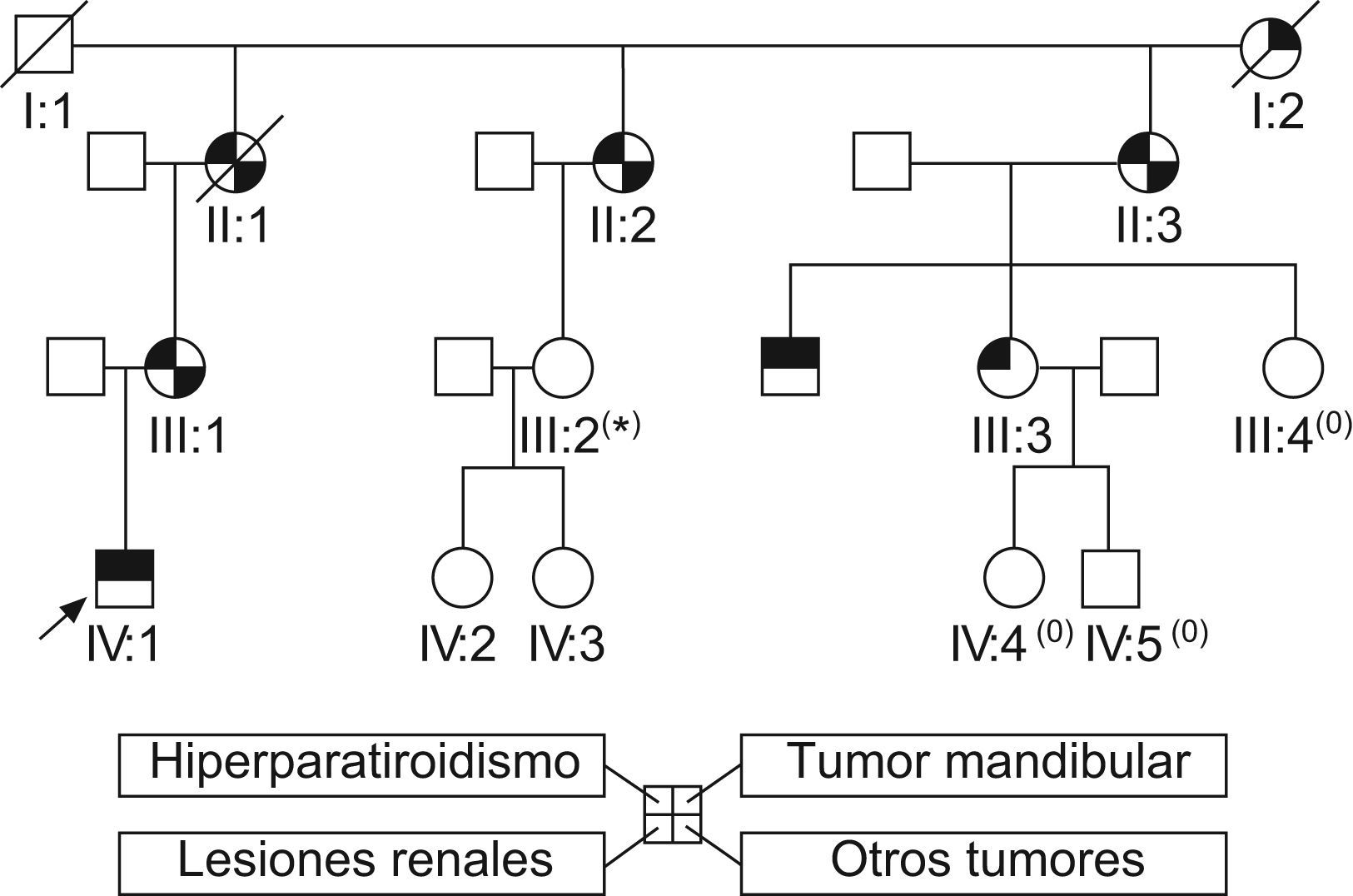
El paciente fue diagnosticado de HPT por CP en el contexto de un síndrome de HPT familiar tumor mandibular. El estudio genético del caso índice detectó una mutación heterocigota en el exón 2 del gen HRPT2 (c.165C>A p.Tyr55X), ya descrita y que puede ser considerada como responsable de la enfermedad. El estudio de los familiares nos permitió identificar a varios miembros afectos de HPT y de algún otro tumor benigno (ningún fibroma osificante ni lesión renal), hasta ese momento asintomáticos. En varios familiares asintomáticos el estudio genético está pendiente en el momento actual, porque residen en el extranjero. La historia clínica nos permitió realizar el árbol de la figura 2.
: pendiente de resultado de estudio genético. (0): pacientes no estudiados, residentes en el extranjero. II: 1) Adenosarcoma endometrial. II: 2) Carcinoma papilar de tiroides; carcinoma ductal infiltrante de mama; carcinoma basocelular; miomas uterinos. II: 3) Hiperplasia nodular de tiroides. III: 1) Adenoma paratiroideo atípico; hiperplasia nodular de tiroides. III: 3) Carcinoma papilar de tiroides. IV: 1) Dos carcinomas paratiroideos.")
Árbol genealógico. La flecha identifica el caso índice. Las líneas cruzadas identifican a los miembros que han fallecido. (*): pendiente de resultado de estudio genético. (0): pacientes no estudiados, residentes en el extranjero. II: 1) Adenosarcoma endometrial. II: 2) Carcinoma papilar de tiroides; carcinoma ductal infiltrante de mama; carcinoma basocelular; miomas uterinos. II: 3) Hiperplasia nodular de tiroides. III: 1) Adenoma paratiroideo atípico; hiperplasia nodular de tiroides. III: 3) Carcinoma papilar de tiroides. IV: 1) Dos carcinomas paratiroideos.
Hasta un 5% de los HPT son hereditarios y se presentan en el contexto de síndromes endocrinológicos (neoplasia endocrina múltiple [MEN]1, MEN2A y el síndrome del HPT familar tumor mandibular [HPT-JT]) o en forma de HPT hereditario aislado.
El HPT-JT (tumor mandibular) es un trastorno autosómico dominante de penetrancia incompleta, caracterizado por desarrollar tumores paratiroideos (con frecuencia, adenomas atípicos o carcinomas), lesiones fibro óseas (fibroma osificante) de la mandíbula o el maxilar, y una variedad de lesiones renales (enfermedad poliquística, hamartomas, tumor de Wilms). Solo se han descrito unas pocas familias afectas de este síndrome. El curso clínico del HPT es más agresivo que el del HPTP esporádico y frecuentemente se afectan más de una glándula paratiroides, de modo sincrónico o asincrónico. Algunos autores han descrito una penetrancia baja del HPT entre las mujeres portadoras1, hecho que no se confirma en la familia aquí descrita. Se ha sugerido que la determinación de la calcemia puede no tener una sensibilidad del 100% para detectar la presencia de patología paratiroidea en los portadores de esta mutación, puesto que se han descrito casos de adenomas atípicos y carcinomas paratioideos en sujetos normocalcémicos2. El tratamiento quirúrgico del HPT en este contexto puede consistir en la resección de la/s glándula/s claramente aumentadas de tamaño, si bien se recomienda explorar las 4 paratiroides3. Si se sospechase la presencia de un carcinoma se ha recomendado la realización de resección en bloque más lobectomía ipsilateral más resección de los tejidos adyacentes4. Hasta un tercio de los pacientes tienen fibromas osificantes de mandíbula, que aparecen generalmente en la adolescencia, son histológicamente distintos de los tumores pardos del HPT y no desaparecen tras la curación del HPT. Además de la asociación con lesiones renales (hamartomas, quistes renales y tumor de Wilms), se ha sugerido que otros tumores (pólipos uterinos, adenocarcinoma de colon, carcinoma papilar de tiroides y adenomas tiroideos) pueden formar parte del síndrome HPT-JT5. De hecho nosotros encontramos en nuestros pacientes una alta incidencia de otros tipos de tumores. También se ha descrito la asociación de tumores no paratiroideos benignos y malignos (cáncer de mama, lipomas, cáncer colorectal, cáncer papilar de tiroides, melanomas y angiomiolipomas) en pacientes con HPT familiar aparentemente aislado6.
Alrededor del 1–2% de los casos de HPT esporádicos se deben a un CP7,8. El CP tiene mayor incidencia en determinados HPT hereditarios; en el síndrome HPT-JT hasta el 15% de los casos de HPT se deben a CP. Las mutaciones inactivadoras del gen supresor de tumores HRPT2 tienen un papel central en la patogenia de los CP, tanto esporádicos como en el contexto del síndrome de HPT-JT9,10 y también se han descrito en alguna familia con HPT familiar aparentemente aislado2,6. Diversos estudios evidencian una alta especificidad y sensibilidad de las mutaciones de este gen para el diagnóstico de carcinoma, lo que lo convierte en un test potencial en el diagnóstico diferencial de neoplasias paratiroideas atípicas, con histología equívoca o no concluyente. Las mutaciones somáticas de otros genes también podrían jugar un papel en la patogénesis del CP. La hipercalcemia es la causa principal de morbilidad y mortalidad por carcinoma de paratiroides. El diagnostico de carcinoma no suele estar disponible antes de la cirugía. El diagnóstico histológico es a menudo difícil. El tratamiento de elección del CP es la cirugía. Los tratamientos no quirúrgicos del CP tienen resultados decepcionantes. Las recurrencias o metástasis también se pueden tratar quirúrgicamente, a menudo con fines paliativos con el objetivo de controlar la hipercalcemia. El cinacalcet puede ser eficaz para reducir el calcio sérico en este contexto11. El CP suele ser un tumor de crecimiento lento, pero alrededor de un tercio de los pacientes presentan un curso rápidamente progresivo. Es frecuente el desarrollo de recidiva local en el cuello después de la cirugía aparentemente exitosa.
En resumen, presentamos una nueva familia afecta del síndrome HPT-JT. A diferencia de otras familias previamente descritas, nosotros no encontramos una baja penetrancia del HPT entre las mujeres afectas. Sí hemos encontrado alta incidencia de otra serie de tumores benignos y malignos en los afectos de más edad.