


Type 1 pseudohypoaldosteronism (PHA-1) was first described in 1958 by Cheek and Perry.1 It is a rare syndrome of aldosterone unresponsiveness, expressed in two forms: renal PHA-1 and systemic PHA-1.2,3 Renal PHA-1 results from autosomal dominant mutations in the kidney mineralocorticoid receptor. As the mineralocorticoid resistance is limited to one organ, the phenotype is milder and often improves spontaneously due to proximal nephron maturation. Systemic PHA-1 results from autosomal recessive mutations in the genes encoding α, β and γ subunit of epithelial sodium channels (ENaC) that exist in multiple organs (kidney, colon, lung, salivary and sweat glands), and therefore the phenotype is severe. Symptoms manifest during the first week of life and require prolonged hospitalizations. Salt-wasting episodes recur frequently and the patients need lifelong high-salt therapy. The mortality rate is high, especially during the neonatal period.
In both forms, diagnosis is established by the presence of high levels of serum aldosterone and plasma renin activity associated with findings typical of hypoaldosteronism (hyponatremia, hyperkalemia and metabolic acidosis).
Herein we describe the evolution of a previously reported case of systemic PHA-1 due to homozygous mutation in intron 3 of the SCNN1A gene (c.1052+2dupT)4 and our therapeutic approach.
Male child, born at full term with birth weight of 3010g (10–25th percentile). There was no parental consanguinity. His 11-year-old sister had Chediak–Higashi syndrome. He was admitted in the Emergency Room at the tenth day of life with hypovolemic shock, severe hyponatremia (125mEq/L), hyperkalemia (>10mEq/L) and metabolic acidosis (pH 7.28, pCO2 48.9mmHg, HCO3 22.6mmol/L, BE −4mEq/L). He received normal saline to correct dehydration and calcium gluconate, sodium bicarbonate, nebulized salbutamol, insulin infusion and rectal cation-exchange resin (sodium polystyrene sulfonate) to control hyperkalemia. Initially a clinical diagnosis of congenital adrenal hyperplasia was made and he started hydrocortisone and fludrocortisone. Later, an endocrinological study revealed normal levels of serum cortisol, ACTH, 17-hydroxyprogesterone, DHEAS and thyroid function, but high levels of serum aldosterone (1750ng/dL: range 7-184ng/dL) and plasma renin activity (70ng/ml/h; range 0.4-1.9ng/ml/h), a diagnosis of PHA-1 was made. Glucose and insulin infusion, calcium gluconate, sodium bicarbonate and nebulized salbutamol were tapered and stopped, and potassium was controlled with high dose of cation-exchange resin. Sodium balance was achieved with 35mEq/kg/day of sodium chloride. During hospitalization, he presented recurrent episodes of tachypnea and fever mimicking respiratory infections but without identifiable bacterial infection. He was discharged at 5 months of age on oral saline (33mEq/kg/day) and cation-exchange resin (1g/kg, six times/day).
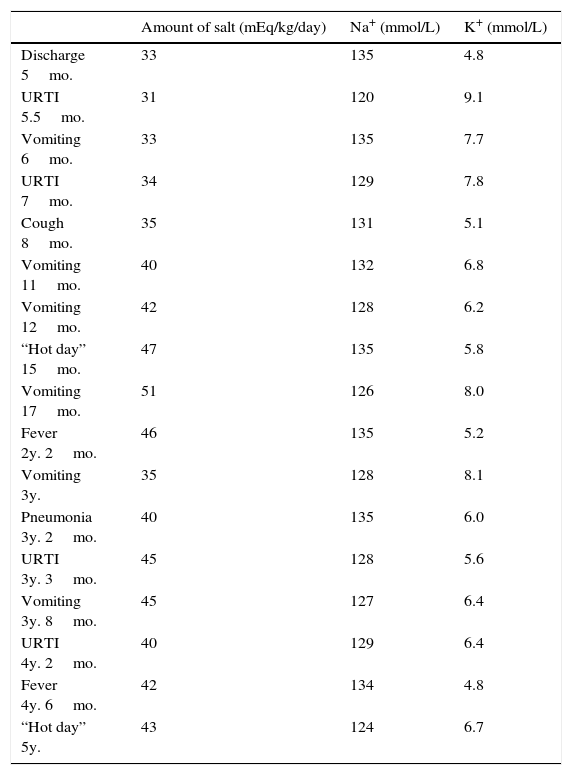
He had frequent follow-up pediatric endocrinology consultations with good therapeutic adhesion. Nevertheless recurrences of fluid and electrolyte imbalances appeared (Table 1) and he was admitted several times to the emergency room with hypovolemic shock, requiring intensive treatment, increase of cation-exchange resin and frequent nebulized salbutamol and calcium gluconate. He had several episodes that mimicked recurrent respiratory infections, characterized by cough, tachypnea, fever and wheezing. These respiratory episodes probably occurred due to defective sodium dependent liquid absorption and mucociliary function that increased airway liquid volume and narrowed airways lumen.5 These symptoms became less severe and less frequent with increasing age.
Amount of salt administration and Na+/K+ balance during specific periods of admissions.
| Amount of salt (mEq/kg/day) | Na+ (mmol/L) | K+ (mmol/L) | |
|---|---|---|---|
| Discharge 5mo. | 33 | 135 | 4.8 |
| URTI 5.5mo. | 31 | 120 | 9.1 |
| Vomiting 6mo. | 33 | 135 | 7.7 |
| URTI 7mo. | 34 | 129 | 7.8 |
| Cough 8mo. | 35 | 131 | 5.1 |
| Vomiting 11mo. | 40 | 132 | 6.8 |
| Vomiting 12mo. | 42 | 128 | 6.2 |
| “Hot day” 15mo. | 47 | 135 | 5.8 |
| Vomiting 17mo. | 51 | 126 | 8.0 |
| Fever 2y. 2mo. | 46 | 135 | 5.2 |
| Vomiting 3y. | 35 | 128 | 8.1 |
| Pneumonia 3y. 2mo. | 40 | 135 | 6.0 |
| URTI 3y. 3mo. | 45 | 128 | 5.6 |
| Vomiting 3y. 8mo. | 45 | 127 | 6.4 |
| URTI 4y. 2mo. | 40 | 129 | 6.4 |
| Fever 4y. 6mo. | 42 | 134 | 4.8 |
| “Hot day” 5y. | 43 | 124 | 6.7 |
Mo., months; URTI, upper respiratory tract infections; y., years.
He had an atopic dermatitis-like rash that was probably the result of increased salt-loss through the skin.6
At 18-months-old, he had his first seizure in apirexia. Other six simple febrile seizures occurred. Electroencephalogram and brain magnetic resonance were normal. Analytical monitoring showed transient subclinical hypothyroidism, asymptomatic hypoglycemia and normal ACTH, cortisol, C-peptide, insulin and IGF-1. ACTH stimulation-test was normal.
Medication was provided by nasogastric tube until 2-years-old. Empirically, hydrochlorothiazide was started from 18 –months old until 4 years old (maximum 2mg/kg/day). Fludrocortisone was gradually reduced until 3 years, and later, cation-exchange resin was also decreased until3 years and 8 months old. Sodium supplements ranged from 28 to 55mEq/kg/day.
He had a mild development delay and he was under a stimulation program but presently his Griffiths Mental Development Scale is adequate.
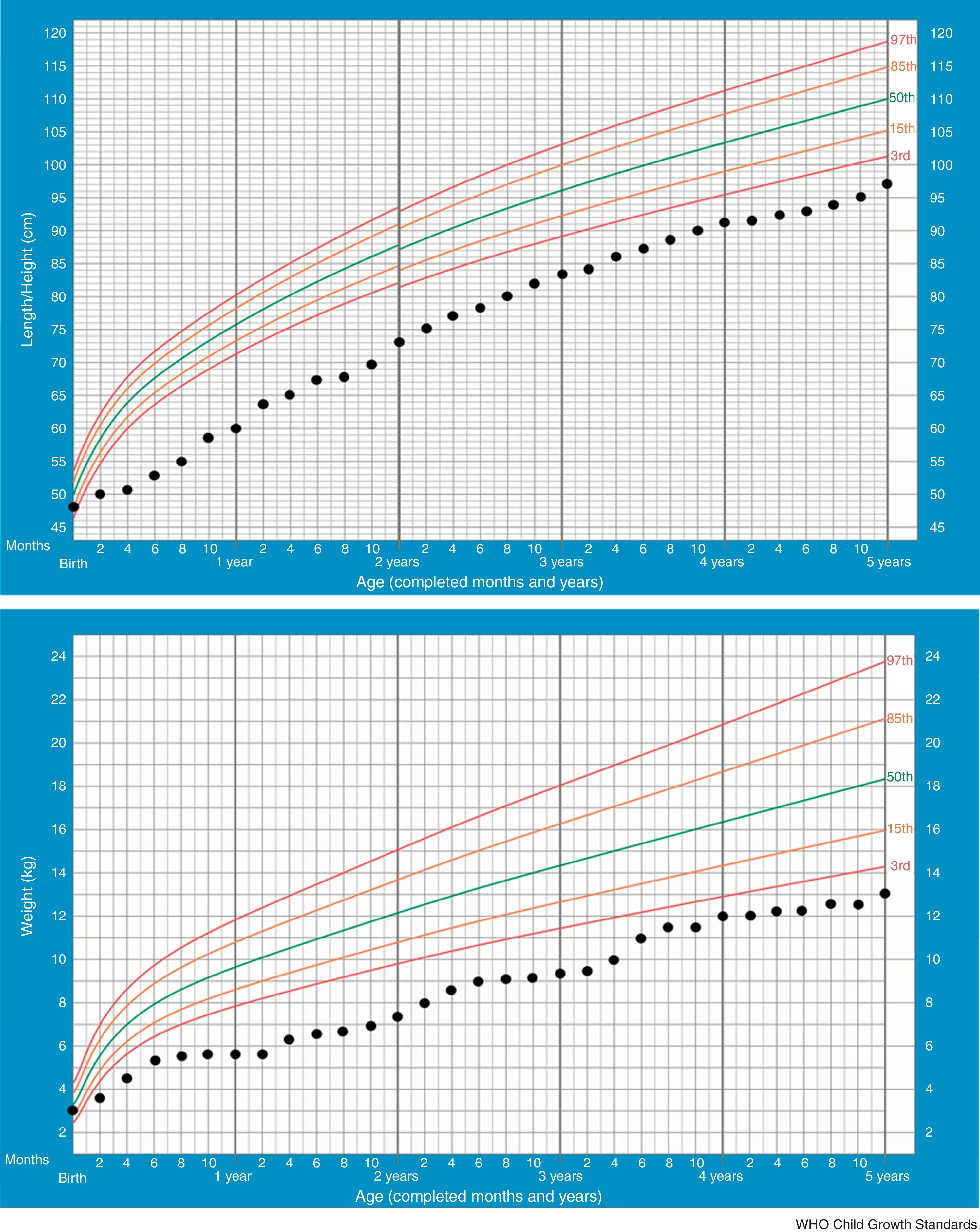
In the majority of cases, growth charts show that patients thrive poorly during the first two years. This is also the most critical period of salt-losing crises. Currently our patient, at 5 –years of age, maintains failure to thrive (height −2.61SDS, weight −3.53SDS) but regular growth velocity (Fig. 1). He only keeps oral sodium supplement.
 from birth to 5 years of age.")
Management of PHA-1 patients is very challenging since there are no evidence-based recommendations. There is scarce literature about hydrochlorothiazide use but it is used to deliver more sodium to the potassium secretory segment. The dosage of sodium per day to establish salt homeostasis varies greatly and has to be readjusted frequently due to changes in body weight. In severe cases this is not enough to prevent salt loss, and death can occur. Moreover, the quality of life of these patients and families is poor: recurrent hospitalizations, large amount of medications, failure to thrive, and susceptibility to infections. An age-dependent trend of amelioration has been reported. The reasons for this improvement are genotype, activity of the truncated ENaC subunit, compensatory increased expression of the NaCl-cotransporter and continued salt supplementation.7,8
The diversity of mutations corresponds to the heterogeneity of clinical phenotypes.3 Mutations are mainly localized on the SCNN1A gene. Most of them are nonmissense mutations, leading to abnormal length protein and a severe phenotype, while missense mutations lead to a normal length protein and are associated with milder phenotypes. In our case, there is a splice site mutation not previously described in the literature; therefore, we do not know its precise effect on the ENaC structure and function. Its location is close to the highly conserved donor splice site of intron 3 and most probably affects the RNA splicing, therefore leading to a grossly abnormal protein.
We would like to share our experience on the difficulties of the management of a patient with genetically proven systemic PHA-1 diagnosed in the newborn period. He fulfilled the criteria listed by Edelheit et al. for severe systemic PHA1: severe salt wasting, frequent hospitalization, recurrent respiratory illness, growth failure, high risk of mortality with requirement of high doses of sodium and cation-exchange resin.9 Our patient had a new splicing mutation in intron 3 and presented a severe phenotype. Despite initial poor prognosis, there was a favorable evolution. Severe presentation in the neonatal period does not indicate life-long severity of the disease.
Ethical declarationsThe authors state that the procedures followed meet the regulations of the responsible clinical ethical research committee, the World Health Organization and the Declaration of Helsinki.
The authors declare that they have complied the protocols of their workplace for publishing patients’ data. The patients included have received sufficient information and have given written informed consent.
FundingNo honorarium, grant, or other form of payment was given to anyone to produce the manuscript.
Conflict of interestThere are no conflicts of interest.