


El mielolipoma adrenal es un tumor benigno, raramente funcionante, que se compone de tejido adiposo y células de extirpe mieloide en diferente proporción. Representa alrededor del 6% de los incidentalomas suprarrenales1 y se encuentra como hallazgo hasta en un 0,2% de los estudios de autopsia2. Existen pocos casos publicados en la literatura de masas mielolipomatosas causantes de síndrome de Cushing3 (SC), por lo que existe la posibilidad de que pase inadvertida la capacidad funcionante que en ocasiones pueden tener estos tumores. Se presenta el caso de un paciente con SC clínicamente manifiesto con un mielolipoma suprarrenal detectado en técnicas de imagen.
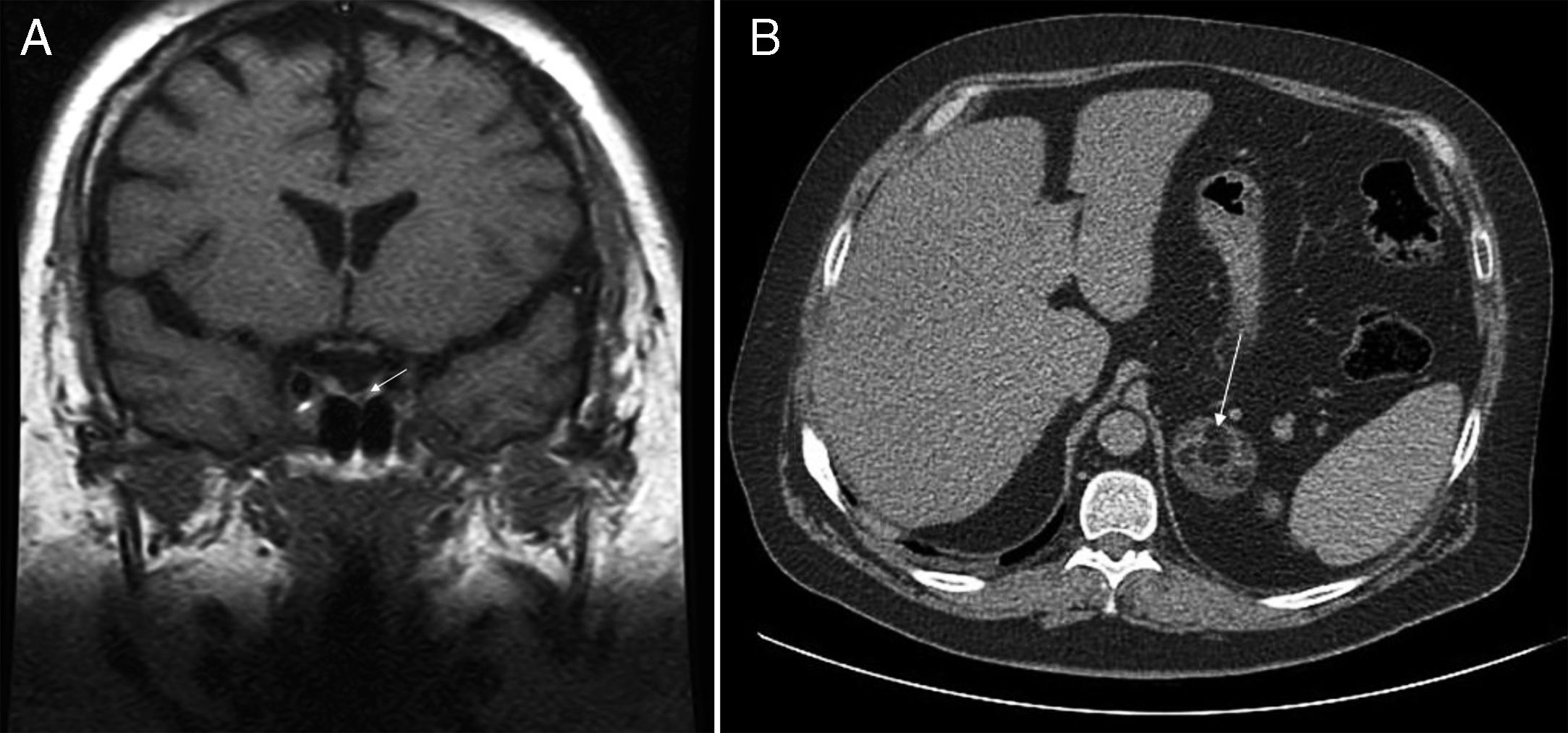
Se trata de un varón de 36 años que acudía a nuestro hospital para control glucémico de su diabetes. El enfermo había sido diagnosticado 10 años atrás de síndrome metabólico con hipertensión arterial, diabetes tipo 2, dislipidemia y obesidad, con mala evolución pese a seguimiento de las recomendaciones higiénico-dietéticas y de la medicación adecuada a su enfermedad. El examen físico revelaba una obesidad central con un índice de masa corporal de 38,6kg/m2, cintura 125cm, tensión arterial 149/109mmHg y rasgos clínicos de SC manifiesto con rubicundez facial, estrías rojo vinosas en abdomen e intensa debilidad muscular. Los niveles plasmáticos basales de ACTH estaban suprimidos (4,5pg/ml; rango normal [RN] 5-45), el cortisol plasmático a las 24h, elevado (15mcg/dl) y el cortisol libre en orina de 24h, elevado (1.218mcg; RN 4,3-176). El cortisol plasmático no se suprimía (16mcg/dl) tras administrar 8mg de dexametasona. La tomografía computarizada (TC) de abdomen mostraba una masa suprarrenal izquierda de 4,5cm de eje mayor, con componente de densidad grasa, sugestivo de mielolipoma (fig. 1A). En la resonancia magnética nuclear (RMN) hipofisaria existía una lesión de 2mm en la región medial izquierda de la hipófisis (fig. 1B). Los niveles plasmáticos de ACTH suprimida y la negatividad de la frenación del cortisol tras altas dosis de dexametasona descartaban el nódulo hipofisario como causa del SC, identificándolo como un incidentaloma del mismo origen. El paciente fue intervenido, extirpando el mielolipoma de la suprarrenal izquierda. El cortisol plasmático postoperatorio fue < 0,1mcg/dl y necesitó tratamiento sustitutivo con corticoides.
 RMN hipofisaria: imagen nodular hipointensa de 2mm de tamaño (flecha), paramedial izquierda, en la secuencia sin contraste realizada en el plano coronal B) TC abdominal: nódulo suprarrenal izquierdo de 4,8cm de eje máximo, con componente de densidad grasa, sugestivo de mielolipoma (flecha).")
A) RMN hipofisaria: imagen nodular hipointensa de 2mm de tamaño (flecha), paramedial izquierda, en la secuencia sin contraste realizada en el plano coronal B) TC abdominal: nódulo suprarrenal izquierdo de 4,8cm de eje máximo, con componente de densidad grasa, sugestivo de mielolipoma (flecha).
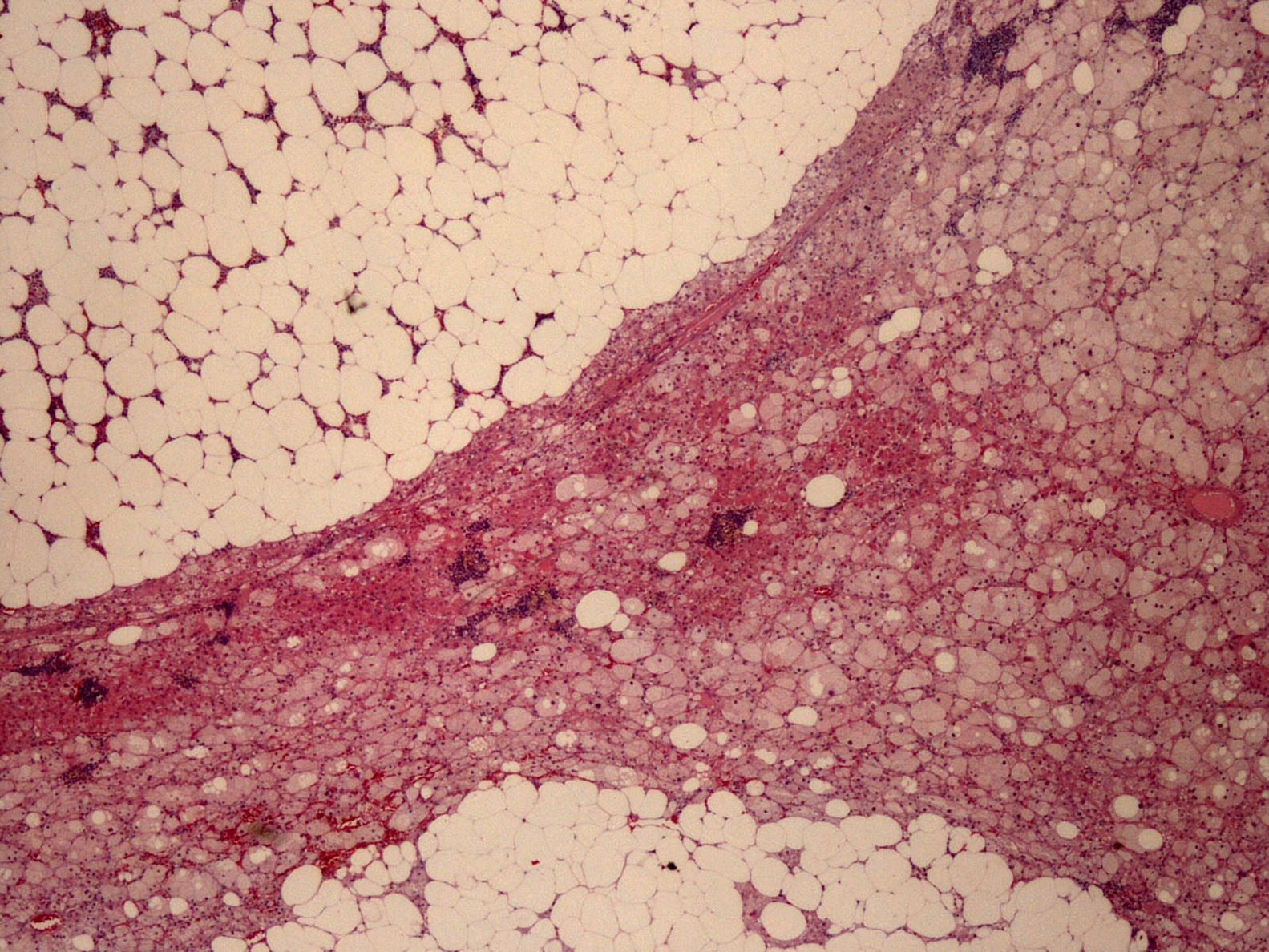
La pieza quirúrgica, recibida como adrenalectomía izquierda, estaba constituida por una formación ovoide de 4,5×4cm en sus dimensiones máximas, de coloración anaranjada y heterogénea, con abundantes zonas pardo-amarillentas. La glándula adrenal no tumoral quedaba comprimida y reducida periféricamente, con un diámetro máximo de 0,4cm. Histológicamente, la neoformación presentaba bordes bien delimitados y se constituía por 2 componentes (fig. 2): el componente de adenoma corticosuprarrenal o adenomatoso (30% de la tumoración) y el componente mielolipomatoso (70% de la tumoración). El primero se disponía formando cordones de células claras y oxifílicas, con citoplasmas a veces vacuolados, debido a su contenido lipídico y núcleos con marcada atipia focal pero escasa actividad mitótica, sin necrosis, cambios mixoides ni invasión vascular; el segundo se formaba por adipocitos maduros y elementos celulares de las 3 series hematopoyéticas, en diferentes fases de maduración.
.")
El estudio inmunohistoquímico, utilizado para confirmar el diagnóstico y para su diferenciación del feocromocitoma, carcinoma suprarrenal o renal entre otras posibles neoplasias, fue positivo para inhibina y melan A (marcadores adrenocorticales), focal con sinaptofisina, pero negativo para cromogranina A (marcador adrenomedular), citoqueratina 7 y CD 10 (marcadores positivos en carcinomas metastásicos de origen renal). Ki 67 se expresó en el núcleo del 1-2% de las células tumorales como signo de su baja actividad proliferativa. El diagnóstico anatomopatológico fue de adenoma corticosuprarrenal con extensa metaplasia mielolipomatosa.
El mielolipoma suprarrenal es un tumor considerado benigno y no productor. No obstante, puede producir un SC grave de varios años de evolución y con fácil resolución tras la adrenalectomía. Dicha asociación la hemos encontrado publicada solo en 10 casos y sus características clínicas difieren del nuestro3, ya que se trataba de SC leves con escasa sintomatología, más frecuentes en mujeres y de corta evolución. En todos los casos la adrenalectomía resolvía la enfermedad. Llama la atención en nuestro paciente la presencia, además, de un incidentaloma hipofisario. Este hallazgo se produce en alrededor del 10% de la población general cuando se realiza RMN cerebral por otro motivo4.
La patogénesis de estos mielolipomas es muy controvertida: la mayoría de las hipótesis se basan en la idea de su origen metaplásico más que en una auténtica neoplasia, como resultado de diferentes tipos de estímulo, incluida la hiperproducción de cortisol, aldosterona o andrógenos por la neoplasia o hiperplasia adrenocortical5. Ha sido también descrito asociado a síndromes de hiperproducción hormonal como feocromocitoma, síndrome de Conn e hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa6–8. A nivel histológico, sería el resultado de la combinación de elementos mixtos de células adrenales con capacidad productiva junto a células mieloides y metaplasia grasa y, al igual que en nuestro paciente, las células adenomatosas (y no los adipocitos ni los precursores hematopoyéticos) serían las responsables de la síntesis hormonal. Por otro lado, estudios hechos con cultivos celulares sugieren un novedoso mecanismo de interacción inmunoendocrina en el que el contacto directo de las células de la corteza suprarrenal con linfocitos T purificados mediarían la estimulación de la secreción de hormonas tales como dehidroepiandrosterona (DHEA) y cortisol9.
Las guías de práctica clínica relativa al manejo de los incidentalomas suprarrenales10 no recomiendan realizar evaluación hormonal de los mielolipomas puros, pero no siempre es fácil distinguirlos de una lesión mixta. Por ello, creemos necesaria la evaluación hormonal de cualquier masa suprarrenal, ya que incluso aquellas con aspecto radiológico de mielolipoma pueden ser secretoras, en especial cuando son heterogéneas en TC o RMN y la clínica así lo sugiere.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.