




Revisar los principios de los microarrays (en concreto array-CGH [a-CGH]), sus beneficios para el diagnóstico prenatal y los desafíos asociados, a través de los trabajos actualmente publicados y aquellos derivados de nuestra experiencia práctica en los dos últimos años.
MétodosRevisión de la literatura y comparación con estudios prospectivos y retrospectivos con array-CGH en casos de gestaciones con elevado riesgo (preferentemente con alteraciones ecográficas).
ResultadosLa tasa de detección de alteraciones genómicas por a-CGH en nuestros casos con alteraciones ecográficas fue de entre el 13,3 y el 15%.
ConclusiónLa combinación de la mayor capacidad de los microarrays en el cribado del genoma humano para la detección de cambios en números de copias, dentro de un único ensayo experimental, así como su mayor resolución técnica en casos de gestaciones de elevado riesgo, podría convertir a los array-CGH, en la próxima década, en una alternativa muy interesante para reemplazar al cariotipo convencional como test diagnóstico de primera línea, en estos casos.
Review the principles of microarrays (in particular array-CGH), their benefits for prenatal diagnosis and the associated challenges, through the currently published works and those derived from our practical experience over the past two years.
MethodsA literature review was carried out, as well as comparisons with prospective and retrospective studies with array-CGH in cases of pregnancies at high risk (preferably with ultrasound anomalies).
ResultsThe detection rate of genomic alterations by array-CGH in our cases with ultrasound anomalies was between 13.3-15%.
ConclusionThe combination of the increased capacity of microarrays in screening of the human genome to detect changes in copy numbers variations, within a single experimental assay, as well as their greater technical resolution in cases of high-risk pregnancies, could lead to array-CGH to be a very interesting diagnostic approach in the next decade to replace the karyotype as a gold standard in such prenatal studies.
En el contexto actual del diagnóstico prenatal, de carácter invasivo, abordamos principalmente su estudio mediante la realización de un cariotipo con bandas-G en cultivo de células fetales (ya sea en biopsia corial, líquido amniótico, o sangre fetal). El cariotipo es capaz de detectar cualquier aneuploidía y/o cualquier gran reordenamiento genómico estructural con un tamaño de entre 5 y 15Mb (de 3 a 10Mb para cariotipos postnatales). La utilización combinada de otras técnicas moleculares, como son la hibridación in situ por fluorescencia (del inglés, FISH), multiplex-dependent ligation probe amplification (MLPA) y quantitative fluorescence-PCR (QF-PCR) han permitido la detección rápida (2-3 días) de las aneuploidías más comunes en muestras prenatales.
Por tanto, bajo cualquier situación que justifique un diagnóstico prenatal invasivo, la pregunta clave que deberíamos tratar de contestar en esta revisión sería: ¿dónde están y en qué situación se encuentran los microarrays en el futuro del diagnóstico prenatal? Para ello, revisaremos el estado actual del uso de esta técnica en el contexto del diagnóstico prenatal, fundamentalmente en gestaciones de elevado riesgo, así como otros aspectos claves, como su asesoramiento genético previo a esta prueba. Intentaremos contestar numerosas inquietudes, entre las que destacamos; a) si es o no factible actualmente recomendar el uso de microarrays en estudios prenatales; b) si las diferentes plataformas pueden o no garantizar de una manera precisa la detección de anomalías cromosómicas en muestras prenatales y si mejoran su tasa de detección versus el cariotipo convencional; c) si por el contario, esta técnica incrementa el número de alteraciones de significancia clínica incierta; d) de cómo abordar las consideraciones éticas, fundamentalmente a través del consejo genético pre- y postanálisis de un estudio por microarray, e) y por último, trataremos de establecer si debemos utilizar o no el microarray como test de primera línea o gold standard en toda muestra prenatal o solo limitarlo a aquellas muestras con elevado riesgo (por ejemplo, en los casos de alteraciones ecográficas).
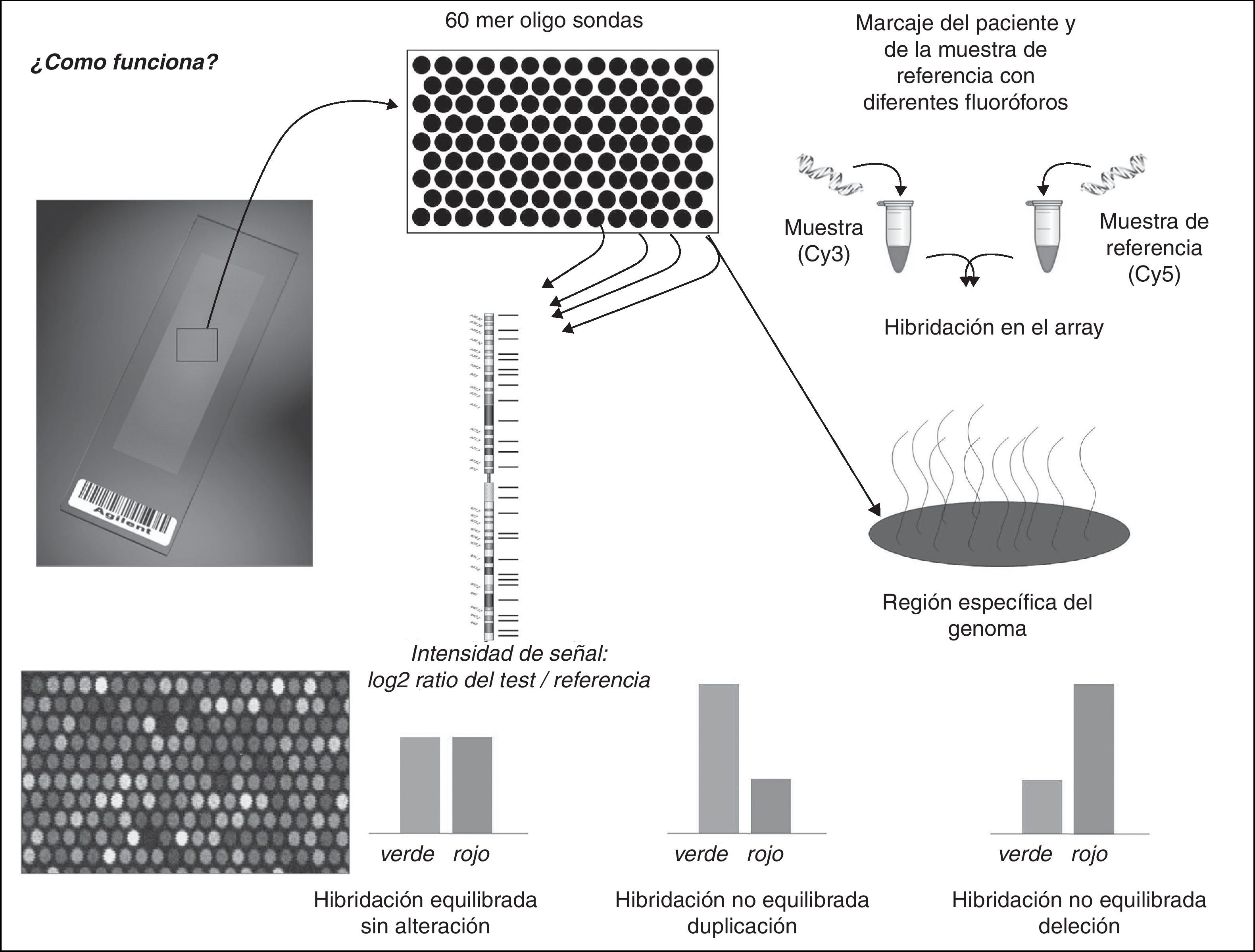
La tecnología de arrays. Tipos de microarraysExisten varios tipos de microarrays (de dosis, de expresión, de metilación, de micro-RNA, etc.), pero los que probablemente están desempeñando y van a desempeñar un papel más determinante en la medicina asistencial son los arrays de dosis (comparative genomic hybridization [a-CGH]), entre los que destacan los arrays basados en bacterial artificial chromosome (BAC), y los arrays de oligonucléotidos. Técnicamente, su mayor utilidad se basa en la aplicación del «CGH» mediante el cual se cohibrida la muestra problema con una muestra control y mediante el uso de fluorescencias distintas en cada muestra, se compara las dosis para la detección de pérdidas o ganancias genómicas (fig. 1).

Por otro lado, los arrays basados en polimorfismos de nucleótido único (single nucleotide polymorphisms [SNP]) tienen una utilidad creciente e importante en la asistencia. Los SNP-arrays aportan información de dosis genómica y adicionalmente de regiones de heterocigosidad/homocigosidad. Además, también es posible encontrar plataformas de oligonucleótidos mixtas para detectar CGH, con sondas de SNP para detectar pérdida de heterocigosidad y disomía uniparental del cromosoma entero, si bien con ciertas restricciones. Finalmente, otro tipo de «array» que utiliza un formato diferente (medio líquido) son los llamados BAC on beads. Una nueva tecnología de alto rendimiento, y rentable para la rápida detección de microdeleciones y aneuploidías fetales, que ya es objeto de un capítulo monográfico en esta revisión.
Ventajas y limitaciones que tiene el uso de microarrays de dosis versus el cariotipo convencionalEl array-CGH (a-CGH) presenta numerosas ventajas tales como; un análisis de alto rendimiento y de gran resolución (capaz de detectar alteraciones cromosómicas entre varias Kbs–1 Mb o superiores), no requiere grandes cantidades de muestra (ADN) para su análisis, lo que evita el cultivo de células fetales en su ámbito prenatal, puede tener un tiempo de respuesta rápido (3-4 días) y el proceso es susceptible de automatización. Adicionalmente, permite la mejor caracterización de las alteraciones citogenéticas. Además, con su uso se pueden analizar objetivamente y de forma simultánea varias regiones genómicas asociadas con trastornos genéticos1–3.
Por otro lado, tenemos que hablar de sus limitaciones: el array-CGH solo detecta cambios de dosis génica, no detecta por tanto, traslocaciones cromosómicas u otros reordenamientos equilibrados como las inversiones, ni mutaciones puntuales. Mediante array-CGH es imposible detectar poliploidías, ya que son cambios globales de material genético, indetectables por los sistemas de normalización de sondas. A veces, un cromosoma marcador tampoco puede detectarse por a-CGH; dependiendo del tamaño, nivel de mosaicismo, su composición y la cobertura de la región cromosómica específica del a-CGH presente en el cromosoma marcador. Además, la cantidad de ADN necesaria para realizar un array-CGH es superior a la necesaria para otras técnicas moleculares, y este debe ser de una calidad adecuada, no estar fragmentado, etc., como puede ocurrir especialmente en especímenes de líquido amniótico. Otra limitación potencial para el uso a-CGH en muestras prenatales es el hecho de la aparición de bajo nivel de mosaicismo, que puede permanecer sin ser detectado. De hecho, la capacidad de detectar este en muestras prenatales por medio a-CGH aún no está bien definida. Varios estudios en muestras postnatales lo establecen en torno al 10%4–10. Nuestros datos en muestras prenatales lo sitúan alrededor de esta misma cifra (con plataformas de BAC11; o en un 13% con plataformas de oligonucleótidos, como el resultado de este estudio). Finalmente, los arrays-CGH de alta densidad pueden detectar un gran número de CNVs, sin un impacto clínico claro a priori conocidas como variant of uncertain significant (VOUS), que pueden dificultar el análisis y/o interpretación de los resultados.
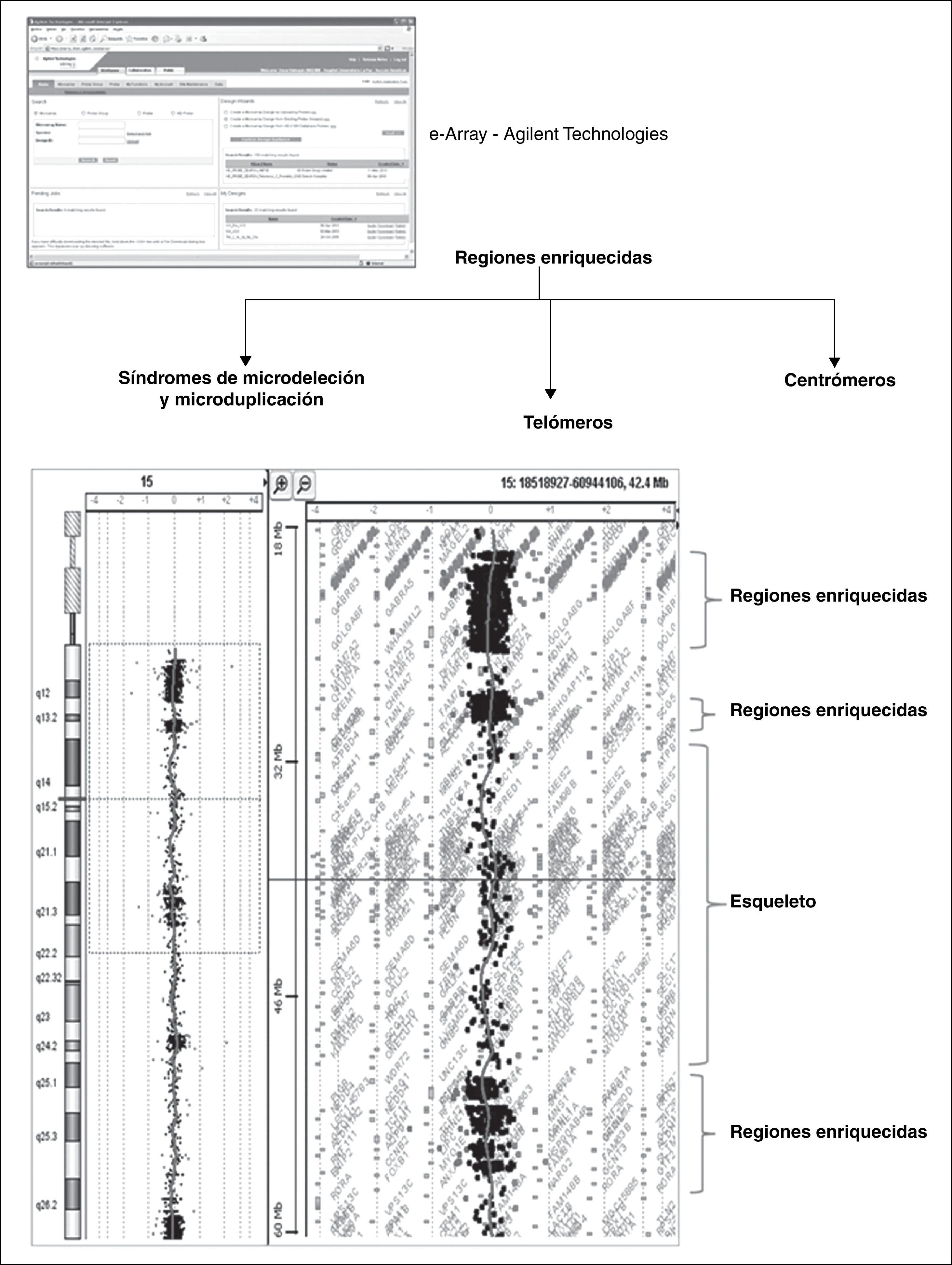
La plataforma ideal de microarrays de dosis para los estudios prenatales. Cobertura y resoluciónLas plataformas basadas en BAC fueron inicialmente las elegidas, no solo en el entorno prenatal sino también en su ámbito postnatal, fundamentado principalmente en la gran ventaja que supone la comprobación de cualquier hallazgo mediante la técnica de FISH. Si bien actualmente la mayoría de los laboratorios y centros de investigación están optando por el uso de array-CGH basados en oligonucleótidos o SNP en lugar de los BAC, ya que estos últimos parecen tender a desaparecer en los formatos comerciales. Así, hablamos de plataformas comerciales o de diseño personalizado y dentro de estos 2 tipos de arrays, destacamos aquellos con cobertura de genoma completo o aquellos dirigidos a regiones de interés patológico (fig. 2). De hecho, la mayor parte de los estudios en muestras prenatales han sido realizados con arrays de BAC12–14, preferentemente en un formato «dirigido», incluyendo regiones específicas del genoma asociadas a síndromes de microduplicación/microdeleción, así como en las aneuploidías más frecuentes. Solo algunos laboratorios muy experimentados, y quizás basados en sus diseños previos realizados y ensayados en versiones anteriores de arrays de BAC que cubren todo el genoma, están empleado arrays de oligonucleótidos con cobertura total y con resoluciones tan altas como 105.000-180.000 sondas (Baudet et al., ESHG, 2011 y Breman et al, ISHG, 2011), para estudios con muestras prenatales. En general, se admite que los arrays basados en oligonucleótidos tienen una capacidad diagnóstica mayor que los arrays basados en BAC (14,83 frente a 9,76%, respectivamente)15. No obstante, con suficiente cobertura de sondas, todas las plataformas actuales de arrays son capaces de proporcionar una sensibilidad suficiente para las pruebas clínicas de array-CGH en el ámbito prenatal. La mayoría de las plataformas clínicas actuales de array-CGH pueden detectar cambios en el número de copias con un límite inferior de la resolución de ∼400Kb lo largo del genoma, lo que representa una resolución al menos 10 veces mayor que el cariotipo. Este nivel de resolución proporcionará un cribado suficientemente fiable para identificar todos los síndromes conocidos de microdeleción y microduplicación recurrentes.
La opinión de los expertos sobre la utilización de los microarrays de dosis en muestras prenatales
En estos últimos años los a-CGH han sido ampliamente introducidos en la rutina clínica postnatal para los estudios de diferentes desequilibrios cromosómicos. De hecho, recientemente se ha publicado un documento de consenso internacional recomendando su uso como primera prueba en lugar de análisis mediante el cariotipo convencional16 para pacientes con retraso mental, malformaciones congénitas múltiples, autismo, entre otros. Por otro lado, una evaluación económica también ha demostrado que en el análisis postnatal, el uso preferencial de microarrays en lugar del cariotipo es rentable económicamente17.
Por otro lado, el Colegio Americano de Ginecólogos y Obstetras (ACOG) en su número 446 de noviembre de 200918 establece una serie de recomendaciones, en las que sigue proponiendo el cariotipo convencional como la principal herramienta para el diagnóstico prenatal. No obstante señala que, el a-CGH en concierto con un adecuado asesoramiento genético, puede ofrecerse como una prueba conjunta en aquellos casos prenatales con hallazgos anatómicos anormales y un cariotipo convencional normal, así como en los casos de muerte fetal con anomalías congénitas y con una cierta incapacidad para obtener un cariotipo convencional. Terminan contemplando al a-CGH como una herramienta de proyección que puede llegar a tener una gran utilidad en el contexto prenatal. Sin embargo, establece que son necesarios estudios adicionales a los que ya estaban en marcha para determinar plenamente su utilidad y sus limitaciones.
En España se está estableciendo un Documento de Consenso para el Uso Clínico de array-CGH a través de un grupo de trabajo promovido por el Instituto Roche (en revisión). El objetivo de este grupo de trabajo es redactar un documento que analice objetivamente las evidencias existentes en este ámbito desde el punto de vista científico, tecnológico y de coste-efectividad. Su recomendación en el ámbito prenatal se centra en no establecer de manera categórica si esta técnica va a tener un uso generalizado o no, ni cuándo se puede incorporar a la rutina prenatal. No obstante, su recomendación establece que si este tipo de estudio es ofrecido a una mujer embarazada, el feto debería previamente haber sido evaluado mediante ecografía prenatal, sometido a cribado bioquímico y haber pasado por un asesoramiento genético prenatal previo al estudio de microarrays, asegurándose que la embarazada o la pareja entienda completamente el alcance del estudio; sus beneficios y limitaciones (Dr. P. Lapunzina, comunicación personal).
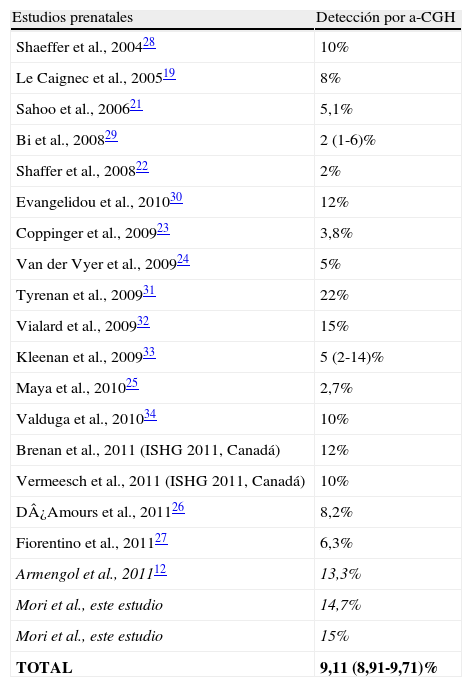
Revisión y discusión de la literatura en el uso de los microarrays de dosis en casos prenatales con elevado riesgoMientras que la experiencia con el a-CGH en diagnóstico en la población pediátrica es extensa, su uso en el ámbito prenatal es todavía muy limitada12–14. Estos primeros estudios se han centrado tanto en estudios retrospectivos19,20, como prospectivos21–25. A pesar del tamaño relativamente pequeño de las cohortes en estos estudios (25-300 muestras, para un total de 1.112 casos estudiados), estos han establecido que el a-CGH es capaz de detectar un mayor número de anomalías cromosómicas clínicamente significativas que el cariotipo convencional12. Pero el dato claramente destacable se obtenía sobre todo en muestras con un riesgo alto, es decir, aquellas con alteraciones ecográficas y cariotipo normal o en muestras con alteraciones citogenéticas en las que no se había podido caracterizar la alteración de una manera adecuada (el porcentaje pasa del 1,3-5,1% a un 8,2-15,4% (tabla 1)19–34. En ellos también se pone de manifiesto que el a-CGH es capaz de detectar un porcentaje nada despreciable de VOUS, a veces tan significativo como un 12,2%26. Este hecho, de nuevo, documentaba las dificultades de interpretación asociadas con las variaciones en número de copia de significado incierto.
Porcentaje de casos con malformaciones (establecidas principalmente por ecografía) detectados por a-CGH en diferentes estudios prospectivos y retrospectivos en muestras prenatales
| Estudios prenatales | Detección por a-CGH |
| Shaeffer et al., 200428 | 10% |
| Le Caignec et al., 200519 | 8% |
| Sahoo et al., 200621 | 5,1% |
| Bi et al., 200829 | 2 (1-6)% |
| Shaffer et al., 200822 | 2% |
| Evangelidou et al., 201030 | 12% |
| Coppinger et al., 200923 | 3,8% |
| Van der Vyer et al., 200924 | 5% |
| Tyrenan et al., 200931 | 22% |
| Vialard et al., 200932 | 15% |
| Kleenan et al., 200933 | 5 (2-14)% |
| Maya et al., 201025 | 2,7% |
| Valduga et al., 201034 | 10% |
| Brenan et al., 2011 (ISHG 2011, Canadá) | 12% |
| Vermeesch et al., 2011 (ISHG 2011, Canadá) | 10% |
| D¿Amours et al., 201126 | 8,2% |
| Fiorentino et al., 201127 | 6,3% |
| Armengol et al., 201112 | 13,3% |
| Mori et al., este estudio | 14,7% |
| Mori et al., este estudio | 15% |
| TOTAL | 9,11 (8,91-9,71)% |
En cursiva se muestran datos basados en nuestra experiencia.
En general, la mayor parte de estos estudios han sido discutidos en una revisión sistemática y estudio de metaanálisis sobre el uso de a-CGH versus el cariotipo convencional en muestras prenatales12, en los que establecen que el a-CGH es capaz de detectar un promedio de 3,6% (1,5-8,5) de desequilibrios genómicos adicionales en muestras con cariotipo normal, al parecer de una manera independiente de la indicación de referencia. No obstante, este porcentaje aumentaba a un promedio de 5,2% (1,9-13,9) cuando la indicación de referencia era una malformación estructural observada por ultrasonido. Curiosamente, el cariotipo no fue capaz de detectar ningún reordenamiento adicional cuando el resultado del a-CGH era normal12, sugiriendo que las áreas en las que esta tecnología no es informativa (traslocaciones equilibradas, inversiones y una menor sensibilidad para triploidías) no parecían ser tan comunes en una población de referencia, si bien hay que señalar que se trataba de una población muy seleccionada en la mayor parte de los estudios.
Por tanto, de este primer grupo de estudios se establecían fundamentalmente 4 conclusiones: a) que la mayor experiencia en clínica prenatal tenía lugar con los arrays enriquecidos de BAC para clones que cubren o flanquean las regiones de síndromes de microdeleción/microduplicación, y/u otros capaces de detectar aneuploidías; b) estableciendo una mayor tasa de detección de anomalías cromosómicas con a-CGH, tanto en muestras con indicaciones generales como en aquellas con alteraciones ecográficas (si bien en estos últimos casos, los porcentajes de detección son significativamente superiores a los primeros).; c) que aquellas plataformas de cobertura de genoma completo parecían generar un mayor número de CNV y d) así mismo, establecían la necesidad del empleo de cohortes de mayor tamaño, antes de cualquier intento de recomendación para su uso clínico rutinario en diagnóstico prenatal como prueba de primera línea (como se recoge en las recomendaciones de la ACOG, 2009)18.
A partir de estos primeros abordajes, nuevos estudios con cohortes más amplias y arrays de segunda generación se han llevado a cabo es estos 2 últimos años, que dan cuenta de unos 6.000 casos más de manera independiente (90612, 1.03727, 4.07312,27,35, casos respectivamente), o un estudio multicéntrico con más de 4.000 muestras prenatales. De este último, cofinanciado por el Instituto de la Salud de los EE.UU. (NIH), a la espera de ser publicado próximamente, solo conocemos resultados parciales que han sido presentados en diferentes reuniones internacionales (ESHG-2011, ASHG/ICHG-2011). Así, de 1.007 casos prospectivos estudiados en uno de estos centros (Baylor College of Medicine, Houston, Tx, EE.UU.), se establecen alteraciones genómicas en un 7,4% de los casos, de los cuales un 2,4% no hubieran sido detectados mediante estudios prenatales convencionales (Breman et al., ASHG/ICHG-2011). De nuevo, estos porcentajes aumentaban considerablemente cuando las muestras procedían de fetos con alteraciones ecográficas (situándose en torno al 12%). Estos resultados eran similares también en otros estudios prospectivos (alrededor del 6,3% de las muestras totales27, tabla 1). El dato más significativo fue establecer que en el resto de condiciones: edad materna, ansiedad parental o cribado bioquímico, la tasa de detección se cifraba en un 0,6%27. Del conjunto de estos nuevos estudios prospectivos parecía sugerirse una mayor evidencia sobre la viabilidad de introducir a-CGH en la rutina del diagnóstico prenatal como prueba de primera línea. Otra vez, parecía establecerse la necesidad de nuevos estudios multicéntricos, con un mayor número de muestras, la comparación entre ellos, así como la creación de nuevos y actuales documentos de consenso sobre el uso clínico rutinario del a-CGH en el diagnóstico prenatal.
MétodosArray CGH personalizado dirigido (Karyoarray® prenatal v3.0, formato 8x15k)El Karyoarray® prenatal, formato 8x15k, es una plataforma de arrays de oligonucleótidos de diseño personalizado dirigido, que abarca más de 36 regiones clínicamente asociadas con desequilibrios genómicos (tabla 2) en un formato en base de Agilent, 8 pacientes por cristal en un total de aproximadamente 15.000 sondas utilizando la página web de e-array (Agilent Technologies, Santa Clara, CA, EE.UU.).
Listado de regiones y síndromes incluidos en Karyoarray®prenatal 8x15K
| Alagille syndrome (AGS) type I |
| Axenfeld-Rieger syndrome type III |
| Blepharophimosis/ptosis/epicanthus inversus syndrome (BPES) |
| Cat-eye syndrome (Type I) |
| Chromosome 13 |
| Chromosome 18 |
| Chromosome 21 |
| Chromosome X |
| Chromosome Y |
| Cri du Chat region 5p25 |
| DiGeorge syndrome 22q11.21 |
| Kallmann syndrome type 1 |
| Langer-Giedion syndrome 8q24.12 |
| Lissencephaly type 1 and Miller-Dieker syndrome (MDS) |
| Muscular dystrophy Duchenne |
| Pelizaeus-Merzbacher disease |
| Phelan-McDermid syndrome 22q13 |
| Potocki-Shaffer syndrome (11p11.2 deletion syndrome) |
| Prader-Willi/Angelman syndrome 15q12 |
| Rubinstein-Taybi syndrome |
| Smith-Magenis syndrome and Potocki-Lupski syndrome 17p11.2 |
| Sotos syndrome |
| Tuberous sclerosis TSC1 gene |
| Williams syndrome 7q11.23 |
| Wolf-Hirschhorn syndrome 4p16.3 |
| Xq28 RETT syndrome MECP2 duplication syndrome |
| 10q23 microdeletion syndrome |
| 11p13 deletion syndrome |
| 15q13.3 microdeletion syndrome |
| 15q24 deletion syndrome |
| 15q26 deletion/duplication syndrome |
| 16pter deletion syndrome |
| 16q24 deletion syndrome |
| 17q21.31 microdeletion syndrome |
| 18q21.3 microdeletion associated with congenital heart disease |
| 1p36 deletion syndrome |
| 1q43-q44 deletion syndrome |
| 2q37 monosomy |
| 3q29 microdeletion syndrome |
| 5q34 microdeletion associated with congenital heart disease |
| 9q subtelomeric deletion syndrome |
El Karyoarray® formato 8x60k es una plataforma de arrays de oligonucleótidos de diseño personalizado (fig. 2), de genoma completo, que abarca más de 350 regiones clínicamente asociadas con desequilibrios genómicos en un formato en base de Agilent 8x60K36. En su estudio de validación se evaluaron diversos tipos de muestras, incluyendo sangre fetal, líquido amniótico y biopsia corial, entre otras.
La diferentes CNVs se tratan como se describe en Miller et al. (2010). Para la gestión de dichas CNVs, es de extraordinaria ayuda el desarrollo de bases de datos públicas, entre las que destacan la base de datos de variantes del genoma (DGV) y las iniciativas de los consorcios ISCA y DECIPHER.
ResultadosDescribimos nuestra experiencia en el empleo del a-CGH en el marco del diagnóstico prenatal, preferentemente en casos de gestaciones con elevado riesgo.
Estudio con arrays de bacterial artificial chromosomeDurante el año 2011 hemos presentado las conclusiones de un estudio multicéntrico comparativo de utilidad clínica (es decir, con la capacidad de establecer la probabilidad de que una prueba dé lugar a un resultado de la mejora de la salud), comparando los costes, así como diferentes metodologías actualmente disponibles para la detección de anomalías cromosómicas después de un muestreo prenatal invasivo12. El estudio fue planteado en una cohorte de muestras consecutivas (alrededor de 900 muestras, a lo largo de un año, entre febrero de 2009 y marzo de 2010 en muestras del INGEMM de Madrid y el Hospital Vall d¿Hebron de Barcelona), con indicación para el muestreo invasivo prenatal. Estas muestras fueron simultáneamente evaluadas mediante 3 metodologías: 1) cariotipo y QF-PCR), 2) 2 paneles de MLPA (P036 y/o P070 y P245; MRC-Holland, Amsterdam, the Netherlands) y 3) array-CGH personalizado de BAC dirigido a regiones patogénicas. Entre los resultados más destacados, el a-CGH detectó un 6,3% de alteraciones con relevancia clínica, un 32% más que el cariotipo y la QF-PCR juntas. Por el contrario, también estableció alrededor de un 2% de VOUS, una cifra que triplicaba la establecida mediante cariotipo y MLPA juntos. En concordancia con estudios previos, el a-CGH también obtuvo una tasa de detección de alteraciones superior en muestras de gestaciones que presentaban alteraciones ecográficas (13,3%)12, aunque también fue significativa en gestaciones a priori consideradas de riesgo bajo (1,7 y 4,0%, para casos con ansiedad y edad materna, respectivamente)12.
Del conjunto de este y de otros estudios prospectivos anteriores parecía establecerse que la aplicación de a-CGH en el diagnóstico prenatal requiere de una gran profesionalidad, precisión y sutileza, porque incluso la detección de una variante familiar puede provocar una gran ansiedad en las parejas. Por tanto, descartar CNV insignificantes o poblacionales y/o resultados inciertos parecía fundamental en este contexto. Con esta idea desarrollamos un diseño de un array de oligonucleótidos a medida, basados en muestra experiencia previa en el ámbito postnatal. En esta nueva situación es importante señalar que, antes de introducir una nueva tecnología, cada laboratorio debe determinar la validez de la herramienta de arrays, así como de todo el proceso tanto analítico como clínico. Para ello, propusimos un estudio de validación, así como diferentes estudios retrospectivos y prospectivos con nuestras herramientas de a-CGH a medida. Estos procesos de validación son parte del modelo de gestión de procesos que está bajo la certificación ISO/9001:2008 (en su apartado Diseño) de AENOR del INGEMM (certificación obtenida en octubre de 2011).
Estudio prospectivo y retrospectivo con microarrays de dosis dirigidos de oligonucléotidos en muestras prenatalesUna consideración importante al seleccionar una plataforma a-CGH es también el precio, especialmente cuando se estudia una amplia serie de muestras. Elegimos por tanto, en función de la necesidad de eliminar CNV benignas en nuestra población y del precio, un diseño personalizado de a-CGH de oligonucleótidos para 8 muestras en un formato de 15.000 sondas (8x15K; Karyoarray® Prenatal v3.0) en base de la compañía Agilent Technologies (Santa Clara, CA, EE.UU.). Un diseño dirigido a cubrir diferentes síndromes de microdeleción/microduplicación recurrentes conocidos (tabla 2), y con una cobertura preferentemente en regiones teloméricas y centroméricas. Su resolución media era de un oligonucleótido cada 10Kb en las regiones de máximo interés. En el resto del array (backbone), la resolución media era de 500Kb, lo que permite detectar pérdidas o ganancias de material genómico de más de 30-50Kb, en regiones de interés o de 1,5-2,5Mb para el resto del array, respectivamente (en función de utilizar entre 3 y 5 sondas consecutivas para considerar una alteración en el número de copias).
En la validación inicial de esta herramienta se usaron de una manera retrospectiva y ciega 50 muestras (33 casos establecidos como patológicos por técnicas convencionales y 17 muestras aparentemente normales) de líquido amniótico, biopsia corial o sangre fetal, antes de iniciar un estudio prospectivo en 24 muestras seleccionadas con alteraciones de alto riesgo (fundamentalmente, aquellas con marcadores ecográficos) y cariotipo normal. En esta validación se incluyeron preferentemente muestras con diferentes alteraciones en síndromes de microdeleción/microduplicación conocidos, así como diferentes tipos de aneuploidías.
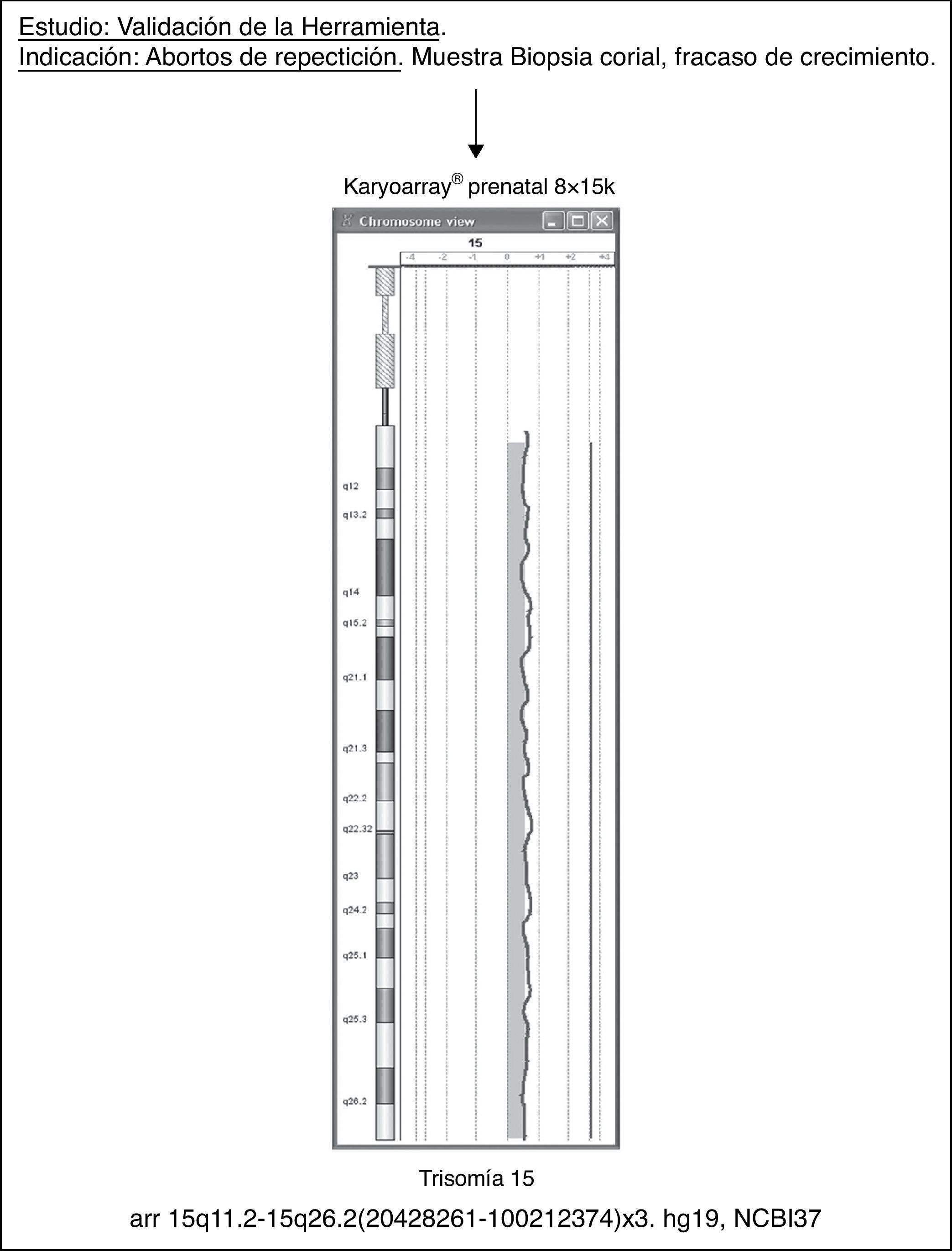
En general, los resultados de la validación con esta plataforma dirigida no fueron demasiado alentadores. De hecho, 5 de las muestras (5/48, 10,41%) no pudieron ser evaluadas, mostrando parámetros de calidad como el derivative log-ratio spread (DLRS) muy elevados y una gran dispersión de las sondas, probablemente como consecuencia de una deficiente calidad del ADN. En otra muestra, el array no fue capaz de detectar una alteración: una poliploidía 69XXY (1/43, 2,33%). No obstante, el a-CGH fue capaz de concretar un caso que no pudo crecer por estudio convencional. Se trataba de una muestra de biopsia corial con fracaso de crecimiento del cultivo en una paciente con abortos de repetición, en la que se estableció una trisomía total del cromosoma 15 (1/43; 2,33% adicional, fig. 3). Durante esta validación, al margen de algunos artefactos claramente visibles, la herramienta mostró un índice de detección de CNV adicionales de 1,33 promedio/muestra para las muestras controles (sin alteración previa) y 1,51 promedio/muestra para las muestras patológicas, siendo muchas de ellas alteraciones benignas. De hecho, la media del valor de los DLRS fue de 0,307 (un valor límite de aceptación para el uso de este tipo de herramienta).

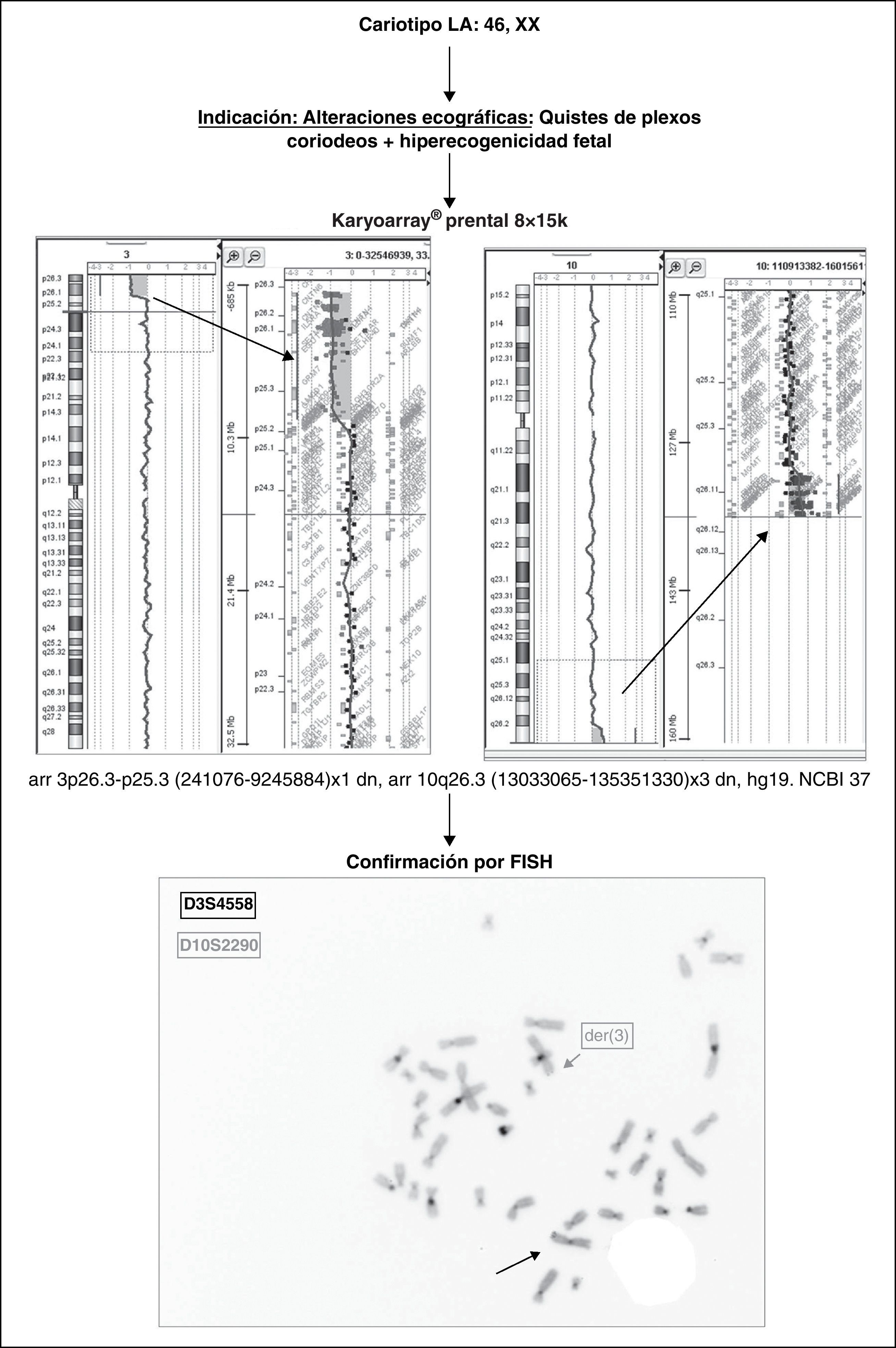
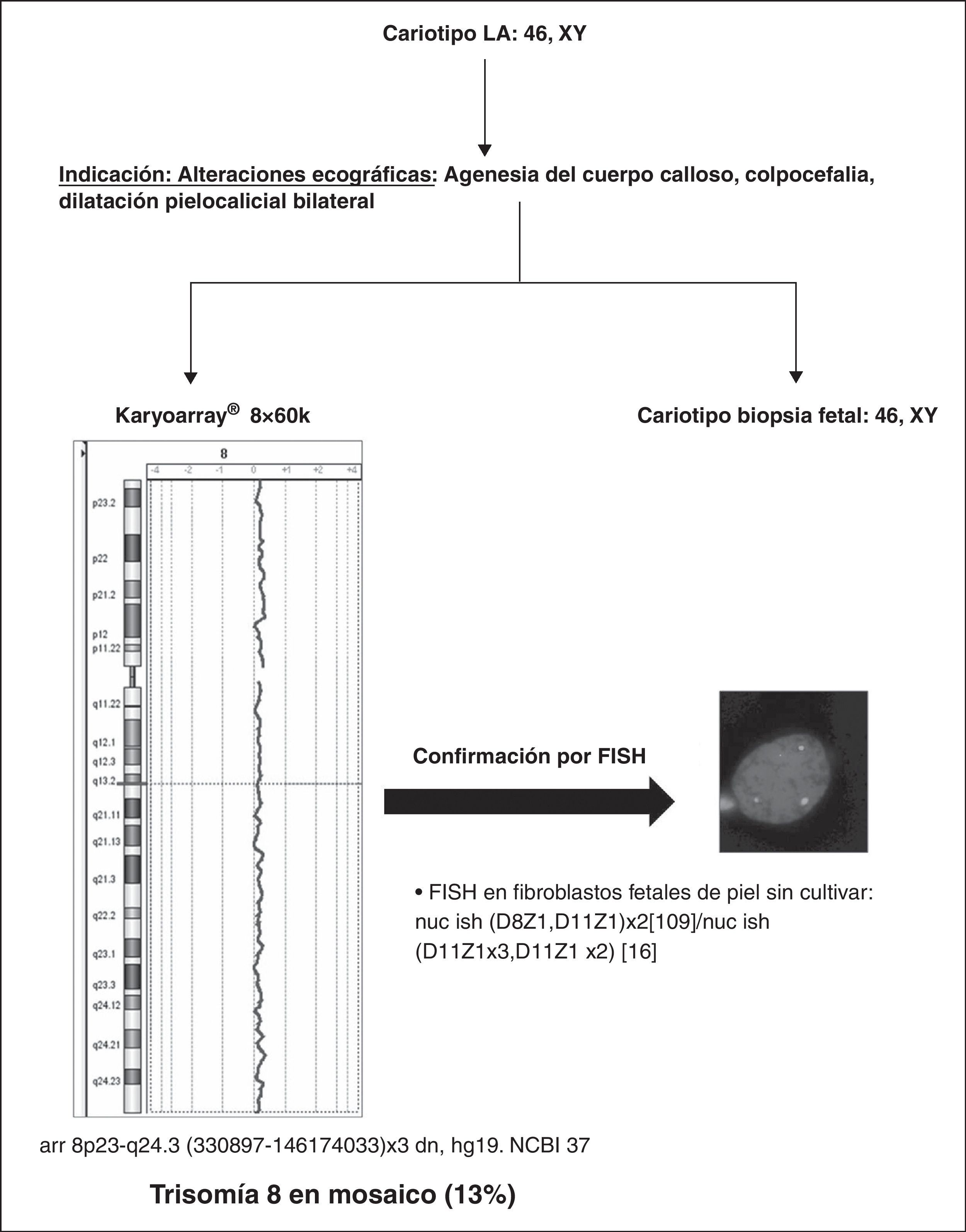
Por otro lado, con un abordaje experimental posterior quisimos establecer su valor pronóstico estableciendo un estudio prospectivo de 3 tandas independientes de 8 muestras prenatales, para un total de 24 casos, preferentemente con anomalías ecográficas y cariotipo normal, repartidas de la siguiente forma: 19 fetos con alteraciones ecográficas, un feto por edad materna, un cribado bioquímico positivo y 3 con ansiedad materna. En términos generales, los resultados mostraron, como anteriormente, unos DLRS altos, con un promedio de 0,313 de las cuales; 7 muestras no pudieron ser analizadas (29%). No obstante, en el total de muestras con alteraciones ecográficas (14 al descontar 5 de ellas no analizables) el array fue capaz de establecer 2 nuevas alteraciones no observadas por técnicas convencionales (2/14; 14,29%). Se trataba de una trisomía 8 y una trisomía X ambas en mosaico en una poliploidía 69, XXY (que no fue capaz de detectar, no mostrado), y un cromosoma derivado 3p/10q en una muestra con 2 marcadores ecográficos menores (quistes de plexos coroideos e hiperecogenicidad cardiaca fetal) y cariotipo normal, que fue confirmado mediante FISH (fig. 4).

En resumen y como conclusión de este estudio tenemos que establecer que nuestra herramienta dirigida a regiones patológicas en este formato 8x15K no parece ser lo suficientemente estable para algunas regiones cromosómicas, como es el brazo corto del cromosoma 19, entre otros, lo que genera unos DLRs altos, con el consiguiente riesgo de no poder determinar algunas alteraciones cromosómicas (falsos negativos) y/o tener artefactos o falsos positivos; estableciendo una sensibilidad y especificidad de la herramienta poco aceptable desde nuestro punto de vista (≤90%).
Estudio en fetos malformados con cariotipo normal por microarrays de dosis de oligonucleótidos de alta resoluciónDe una manera paralela, realizamos un estudio de investigación retrospectivo en 40 fetos con malformaciones aisladas o múltiples (fundamentalmente, con alteraciones a nivel del sistema cardiaco, sistema nervioso central, esquelético, gastrointestinal, o urogenital), y/o restricción del crecimiento intrauterino detectados por ultrasonido, utilizando un a-CGH personalizado de oligonucleótidos en su formato 8x60K (Karyoarray®, basado en Agilent Technologies). Esta herramienta cubre todo el genoma, y mediante la selección de sondas de oligonucleótidos 60-mer a través de la página de e-array (Agilent, https://earray.chem.agilent.com/earray) incluimos todos los síndromes de microdeleción y duplicación conocidos, así como los telómeros y regiones pericentroméricas (∼5Mb). La validación de esta herramienta ha sido llevada a cabo preferentemente en muestras postnatales36. Su resolución presenta una densidad media de cobertura de 43Kb a lo largo de todo el genoma, dividida de la siguiente manera; 9Kb (entre 1y 30Kb) en las regiones de interés y 175Kb en las demás regiones del genoma. Esta resolución en zonas de interés patológico es semejante a la de un array comercial de 244K de Agilent (∼8Kb). Este chip de oligonucleótidos específico, basado en nuestra experiencia clínica y de laboratorio, abarca más de 350 regiones clínicamente asociadas con desequilibrios genómicos36. Para algunos casos se empleó un array comercial de Agilent 4x44K, mientras la validación de Karyoarray® estaba disponible (7 casos). Los resultados son claros: se encontraron 6 alteraciones cromosómicas patogénicas, un 15% (6/40 casos), así como un total de un 10% de variantes polimórficas (4/40). Esta alta tasa de detección de desequilibrios cromosómicos obtenida en este estudio (15%) muestra la eficacia del a-CGH para identificar anomalías cromosómicas clínicamente importantes en fetos con anomalías ecográficas y cariotipo normal. Un ejemplo de este estudio se muestra en la figura 5; el KaryoArray® detectó una trisomía completa del cromosoma 8 en mosaico (13%), en un feto con alteraciones ecográficas (agenesia del cuerpo calloso, colpocefalia, y dilatación pielocalicial bilateral) y con cariotipo en líquido amniótico y tejido fetal normales. No nos cabe duda de que el uso de este tipo de herramienta puede tener un impacto importante sobre el asesoramiento genético en gestaciones con anomalías fetales.

Estos resultados y otros factores entre los que destacamos la mejora de las bases de datos de CNVs y nuestra propia base de datos poblacional, así como nuestra mayor experiencia en el contexto postnatal (más de 1.000 KaryoArray® en nuestra rutina clínica durante este último año y medio), nos hace plantear la posibilidad de implementar el uso de nuestra herramienta postnatal, KaryoArray® (8x60K) en nuestra rutina prenatal invasiva, por el momento, con un estricto criterio de selección de las muestras. La figura 6 muestra 2 alteraciones detectadas por este abordaje; en el estudio de una muestra fetal de una pareja con abortos de repetición, mostrando una trisomía total del cromosoma 22; y una deleción intersticial del brazo corto del cromosoma 6 en una muestra con alteraciones ecográficas que hacían sospechar una cardiopatía (doble salida en ventrículo derecho con CIV, posible comunicación interventricular moderada) y con cariotipo normal.
Discusión
Los microarrays basados en CGH han sido recientemente introducidos en la rutina clínica de pacientes pediátricos con anomalías congénitas, déficits cognitivos, retrasos en el desarrollo, anomalías de crecimiento o problemas de comportamiento (revisado por Miller et al., 2010). De hecho, se están convirtiendo en una herramienta esencial de diagnóstico clínico rutinario y poco a poco están reemplazando a los métodos citogenéticos en el entorno postnatal, no solo por ser capaces de detectar nuevos reordenamientos genómicos, sino también por su capacidad de descubrir nuevos genes y su asociación con la enfermedad16,37,38. Sin embargo, la aplicación de los array-CGH en diagnóstico prenatal se realiza tan solo en unos pocos laboratorios en todo el mundo ¿Por qué esta situación? En principio, no existen barreras técnicas que puedan impedir la introducción del a-CGH en el ámbito del diagnóstico prenatal. La cuestión es más bien cómo se llevará a cabo su implementación y sobre todo, hacia qué muestras prenatales estará principalmente dirigido. La presencia de CNV con un significado clínico incierto, particularmente aquellas que muestran una penetrancia reducida o expresión variable, los hallazgos accidentales y aquellos no relacionados con las anomalías fetales observadas parecen complicar su uso en este contexto.
Lo cierto es que la implementación en nuestra rutina diagnóstica prenatal de los arrays es una realidad tangible que debemos incorporar de forma gradual para beneficio de los pacientes y familias que consultan en los Servicios y Departamentos de Genética. Como toda nueva técnica es inevitable que surjan controversias acerca de su aplicación clínica potencial. Para los clínicos, la dificultad para explicar la implicación patológica de ciertas CNV a los pacientes es evidente, lo que hace que muchos nieguen la utilización del a-CGH en el diagnóstico clínico de muestras prenatales. En este sentido, los a-CGH personalizados y diseñados a medida, ya sean de genoma completo o dirigido, han sido ampliamente desarrollados para disminuir esta incertidumbre, buscando un equilibrio entre la gran cantidad de información que pueden proporcionar este tipo de herramientas y la limitada información que nos proporcionan otras técnicas convencionales en diagnóstico prenatal.
Nuestra experiencia en el contexto prenatal basada en la utilización de diferentes tipos de plataformas de arrays (de BAC u oligonucleótidos, ya sean dirigidas o de alta resolución cubriendo todo el genoma), y preferentemente en muestras con alteraciones ecográficas, nos hace considerar que en efecto, el a-CGH debería ser una opción de primera línea para el diagnóstico prenatal invasivo en gestaciones de elevado riesgo, en lugar de la actual combinación de métodos convencionales: QF-PCR, FISH, MLPA y cariotipo. De hecho, del conjunto de los 3 tipos de estudios que hemos realizado en estos 2 últimos años, hemos establecido una tasa de detección de alteraciones en muestras con elevado riesgo (fundamentalmente alteraciones ecográficas), por medio de a-CGH en torno a un 14% (13,3-15%), tanto para cohortes prospectivas como en población muy seleccionada. Datos de gran concordancia con los casos previamente publicados12 (tabla 1).
Por otro lado, en nuestra casuística de laboratorio en estos últimos 2 años hemos establecido que el porcentaje de alteraciones cromosómicas establecido por métodos convencionales en muestras con alteraciones ecográficas era un 2,55 y 4,16% (para los años 2011 y 2010, respectivamente). Quizás, el dato más destacable era que los casos con alteraciones ecográficas representaron el 48,7 y el 53,7% del total (para los años 2011 y 2010, respectivamente). Estos números son probablemente muy similares a los de otros centros de nuestro país. De hecho, el Instituto Dexeus de Barcelona, en un balance recientemente publicado sobre el uso de técnicas invasivas prenatales, a lo largo de más de 9.000 muestras en los últimos 10 años, ha establecido que el mayor valor predictivo positivo es para aquellas muestras referidas al laboratorio por alteraciones ecográficas (alrededor del 39%). Si comparamos con otros estudios previos, los datos son muy variables pero reflejan el gran peso de estos casos sobre su casuística total (4,1-48,3%)21,23,25,27,39,40. Unos datos, significativamente muy inferiores en casos de riesgo bajo, como la ansiedad materna (0,45%)41. Finalmente, habría que reseñar que el estudio retrospectivo de nuestra casuística en los 2 últimos años establecería en el 1,21% el porcentaje de alteraciones que no hubieran sido, a priori detectadas por un a-CGH, al tratarse de traslocaciones citogenéticamente equilibradas o inversiones. Un dato acorde con otras casuísticas (0,72%)41.
Si el CGH debe ser utilizado en diagnóstico prenatal como una prueba de primera línea ya ha sido ampliamente debatido42-44. De hecho, los resultados presentados en este estudio, junto con las experiencias anteriores, sugieren que podría ser ya aceptable ofrecer a-CGH a las mujeres a las que se les han realizado pruebas prenatales invasivas de rutina, por lo menos, simultáneamente con el cariotipo convencional, si bien, más estudios prospectivos en esta área, con mayores cohortes, serán vitales para dilucidar finalmente el papel que esta técnica llegará a desempeñar en el diagnóstico prenatal, incluyendo si puede reemplazar el uso de cariotipo estándar. En cualquier caso, antes de reemplazar una técnica por otra, estas deben coexistir siendo complementarias unas con otras hasta su total validación y en su caso su posible reemplazo. La oferta de a-CGH a una población prenatal más amplia podría probablemente dar lugar a una mayor tasa de detección, valorada por este y otros estudios entre el 1,8 y el 6%12. Este hecho podría tener una relevancia mayor si, como la casuística de algunos centros establece, en realidad hasta el 40%22 de las muestras neonatales que requieren consulta de asesoramiento genómico fueron referidas por rasgos dismórficos, una indicación clínica que rara vez es identificada en la población prenatal. De hecho creemos que la minimización del número de VOUS es solo una cuestión de tiempo, probablemente como consecuencia de una mejor descripción de estas variaciones en grandes cohortes. En ese futuro escenario, la utilización de plataformas de alta resolución, con una cobertura de genoma completa podría establecerse como primera elección. La resolución óptima de una plataforma de a-CGH para su uso prenatal aún no ha sido establecida. Parece que los arrays dirigidos son adecuados por su capacidad para identificar CNV en loci conocidos del genoma humano asociadas a enfermedades específicas, pero corren el riesgo de no detectar CNV patógenas no establecidas en estas regiones genómicas particulares. Por otro lado, una plataforma de alta resolución tendrá la capacidad de detectar un mayor número de CNV pero también, un mayor riesgo de obtener más resultados de significado incierto, que incrementan el tiempo y la dificultad de su interpretación y provocan una incertidumbre adicional en las parejas. Se trata por tanto de encontrar un equilibrio entre la resolución, su capacidad de detección, su precio y las VOUS. En este sentido, nuestra plataforma KaryoArray® de genoma completo 8x60K cubre satisfactoriamente todas estas expectativas.
Por tanto, en términos generales parece que el a-CGH nos permite encontrar un mayor número de alteraciones genómicas tanto en el ámbito postnatal16, como en el prenatal12–14, pero supone también un gran número de inconvenientes. Bajo estas circunstancias, la pregunta clave es ¿por qué seguimos interesados en utilizar esta nueva tecnología? En nuestra opinión, son varias las razones que nos pueden llevar a justificar su uso potencial, entre ellas podemos destacar: a) hay que tener claro que en la práctica el diagnóstico prenatal ha estado y está centrada principalmente en diagnosticar la aneuploidía mas frecuente en nacidos vivos, la trisomía 21 (frecuencia 1/700). No obstante hoy conocemos que, de manera conjunta diferentes síndromes de microdeleción pueden tener una frecuencia mayor que el de Down, y adicionalmente, su grado de minusvalía ya sea física o psíquica en algunos de estos síndromes es similar y/o más severo que el síndrome de Down, como son el síndrome de Prader Willi/Angelmann, síndrome de Williams o síndrome velocardiofacial; b) el cariotipo convencional mediante bandas-G presenta también limitaciones, en cuanto a su resolución, al tiempo y a la dependencia de un observador; c) otro aspecto nada despreciable en el contexto prenatal es la necesidad de cultivo celular de media entre 15-20 días un intervalo que representa un momento de gran angustia para la pareja. La rapidez y la precisión son, por tanto, 2 requisitos importantes en el diagnóstico prenatal. La realidad es que existe una clara necesidad de técnicas alternativas, más rápidas, de fácil manejo y con posibilidad de automatización, al menos en embarazos de alto riesgo, pero especialmente cuando hay un plazo legal limitado para la terminación del embarazo. Hoy el a-CGH puede llegar a satisfacer esta serie de recomendaciones; d) la evidencia en estudios postnatales disponible sugiere que los reordenamientos equilibrados verdaderos (sin pérdidas o ganancias) representan solo una pequeña proporción en la población y que los reordenamientos «aparentemente» equilibrados citogenéticamente muchas veces no lo son a nivel del ADN, especialmente en pacientes con un fenotipo anormal (esta cifra se acerca a un 45% de los casos con reestructuraciones)45,46; e) desde 2009 los costes económicos de ambos métodos (a-CGH y técnicas convencionales) se han ido acercando cada vez más; f) fundamentalmente, la mejora, el incremento y la actualización de las bases de datos son de gran utilidad para el análisis e interpretación de los array-CGH; g) aunque los ensayos no invasivos para diagnóstico fetal son un creciente campo de investigación, en la actualidad son solo aproximaciones experimentales47.
En resumen, la mayor capacidad diagnóstica de los microarrays parece que permite superar con mucho estas preocupaciones. Aún así, recomendamos expresamente tener algunas consideraciones adicionales inherentes a la técnica de obligado cumplimiento durante el asesoramiento pretest y postest. Por tanto creemos que, antes de usar a-CGH debemos tener clara la información a proporcionar a las parejas durante el asesoramiento preprueba. Esta ya ha sido ampliamente revisada en estudios previos48. Un ejemplo de los requerimientos necesarios se muestra en el anexo 1. Estas medidas facilitarán el análisis por a-CGH, la resolución de problemas y la ansiedad potencial de las parejas. En este sentido, en numerosas ocasiones se necesitarán muestras de ambos padres, que servirán para tratar de resolver problemas de posible contaminación con células maternas, así como la inmediata caracterización de potenciales CNV familiares.
ConclusionesLos autores revisan los principios de los arrays-CGH, sus beneficios para el diagnóstico prenatal y sus desafíos asociados. Aunque su mayor capacidad diagnóstica les hace superar estas preocupaciones. Basados en nuestra experiencia, recomendamos su utilización en muestras de gestaciones con elevado riesgo, al menos de manera conjunta a los medios convencionales, ya que parece claro que incrementa la detección de anomalías genómicas fetales, son coste efectivos, y bien aceptados por las parejas. Las gestantes bajo este tipo de estudio necesitan tener claras las implicaciones de su uso antes de comenzar el estudio: los hallazgos inesperados de significado incierto, o los accidentales, dificultan su interpretación y pueden generar problemas éticos a las parejas.
Hasta hace poco, la detección prenatal de enfermedades genéticas se dirigía a solo un subconjunto de la población gestante considerada de mayor riesgo. El mayor uso de las ecografías, la mejora en su poder de resolución y experiencia en la seguridad diagnóstica, están permitiendo un cambio de tendencia desde las técnicas citogenéticas hasta las moleculares. Quizás las preguntas que debamos tratar en un futuro próximo de contestar serán: a) ¿hay que ofrecer a-CGH a toda aquella gestante bajo un test invasivo? y b) ¿debemos ofrecer un test invasivo a toda mujer embarazada? Debemos por tanto, estar preparados ante un posible incremento de su demanda en un futuro inmediato, incluso por parte de las propias gestantes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Queremos hacer extenso nuestro agradecimiento a todo el personal del INGEMM que ha hecho posible estos estudios, en especial a Victoria E. Fernández Montaño y a Rosario Sansegundo, así como a todas las parejas que han formado parte en los mismos. Parte de la realización de estos estudios ha sido financiada por los siguientes Proyectos de Investigación: FIS-08/1207; REDES-FIBHULP08/Nevado y FIS ETS 08/90784.
- (i)
Objetivo de prueba. Hay que dejar claro que el a-CGH es una tecnología relativamente nueva que permite análisis de cromosomas a nivel molecular, con una resolución muy superior al cariotipo convencional.
- (ii)
Metodología. Ventajas y limitaciones. Hay que dejar claro que la prueba tiene limitaciones y puede provocar mayor ansiedad en las parejas sobre resultados de importancia clínica incierta, que en la actualidad no pueden resolverse. En muchos de los casos esta tecnología se realizará simultáneamente a la realización de un cariotipo. De hecho, se debe dejar claro si solo se va a informar de los hallazgos más relevantes (como muchos laboratorios), y si se informará conjuntamente sobre los resultados citogenéticos y moleculares.
- (iii)
Logística de obtención de muestras (amniocentesis, biopsia corial, sangre fetal, etc.).
- (iv)
Posibilidad de obtener resultados de significado clínico incierto. Esto supone, por un lado, la necesidad de disponer y analizar muestras parentales en ciertos casos (a priori entre un 10 y un 15% de los casos) y por otro, establecer y explicar la existencia de variantes con expresividad variable impredecibles, así como los hallazgos accidentales. Deberemos por tanto, discutir la relevancia clínica y el pronóstico de la alteración detectada. Considerando la posibilidad de una penetrancia incompleta y expresividad variable, junto con la conveniencia de mayor análisis genético de los padres para determinar si la alteración fue heredada o ex novo.
- (v)
Tiene que haber un consentimiento informado.
- (vi)
Tiene que existir una revisión e interpretación de los resultados, mediante una consulta o asesoramiento genético postest.