




Obesity is a chronic disease, currently recognized as the triggering agent for the development of the metabolic syndrome. It is now accepted that obesity rises from an energy imbalance due to excessive food ingestion and insufficient physical activity. Mitochondria has an important role in energy balance and, interestingly, recent findings have found an association between obesity and mitochondrial dysfunction due to a defective network among regulator proteins such as peroxisome proliferator-activated receptors (PPARs), sirtuins (SIRTs), and PPAR coactivator 1 alpha (PGC-1α). These molecules are currently under extensive research in aims of finding new agents that could be used in the treatment of obesity and metabolic syndrome. The paradox: some nutrients themselves, such as flavonoids, are able to modulate the previously mentioned energy regulators.
La obesidad es una enfermedad crónica que es considerada como el agente detonador para el desarrollo de síndrome metabólico. Se ha aceptado que la obesidad surge de un desbalance energético provocado por ingestión alimentaria excesiva y actividad física insuficiente. La mitocondria tiene un papel importante en tal balance energético e, interesantemente, se ha encontrado una asociación entre la obesidad y la disfunción mitocondrial debido a interacciones defectuosas entre ciertas proteínas reguladoras como los receptores activados por proliferadores de peroxisomas (PPAR), sirtuinas (SIRT) y el coactivador de PPAR (PGC-1α). Estas moléculas se encuentran actualmente bajo una amplia investigación con miras a encontrar nuevos agentes que pudiesen ser utilizados en el tratamiento de la obesidad y del síndrome metabólico. La paradoja: algunos nutrientes, como los flavonoides, son capaces por sí mismos de modular los reguladores antes mencionados.
Nutrition is one of the most influential components of health. An individual lacking of an adequate nutritional status can be diagnosed as being either underweight, overweight or obese. The term “overweight” refers to an excessive ratio between body weight and height – i.e., body mass index (BMI) – such extra weight can come from lean body mass (mainly muscle), fat, or body water. In contrast, in obesity, the surplus body weight comes exclusively from fat.1,2The World Health Organization (WHO) defines obesity as a chronic disease characterized by an excessive and abnormally high proportion of fat mass in the body, to the extent that health can become impaired.2 This disease develops as the result of a complex interaction between environmental and genetic factors. Certainly, there are individuals with rare genetic disorders (e.g., Prader-Willi syndrome, Cohen syndrome, leptin or melanocortin receptor deficiency, etc.), in which excess adiposity develops despite the environment; but such alterations can only explain about 5% of the cases of obesity.3–5 However, in common obesity an obesogenic environment must be present so that the obesity-prone genotype can be expressed.6–9An obesogenic environment is defined as one that enhances fat accumulation as a result of prolonged energy imbalance due to insufficient energy expenditure – scarce physical activity – excessive energy intake – from a hyperenergetic/hypercaloric diet – or both.10,11 The impact that such environment has on the expression of the genetic information is clearly shown, e.g., studies about the Pima Indians: an ethnic group settled in northern Mexico and at the south of the U.S.; hence they are separated only by the border between the two countries. Interestingly, the group settled in the U.S., exhibit a significantly higher rate of obesity than their counterparts in Mexico although they share a similar genetic background. It has been found that “the only” difference between them is energy balance: Mexican Pima have a traditional diet that includes less simple carbohydrates, more fiber and less fat along with moderate to heavy occupational and leisure physical activity.12–16
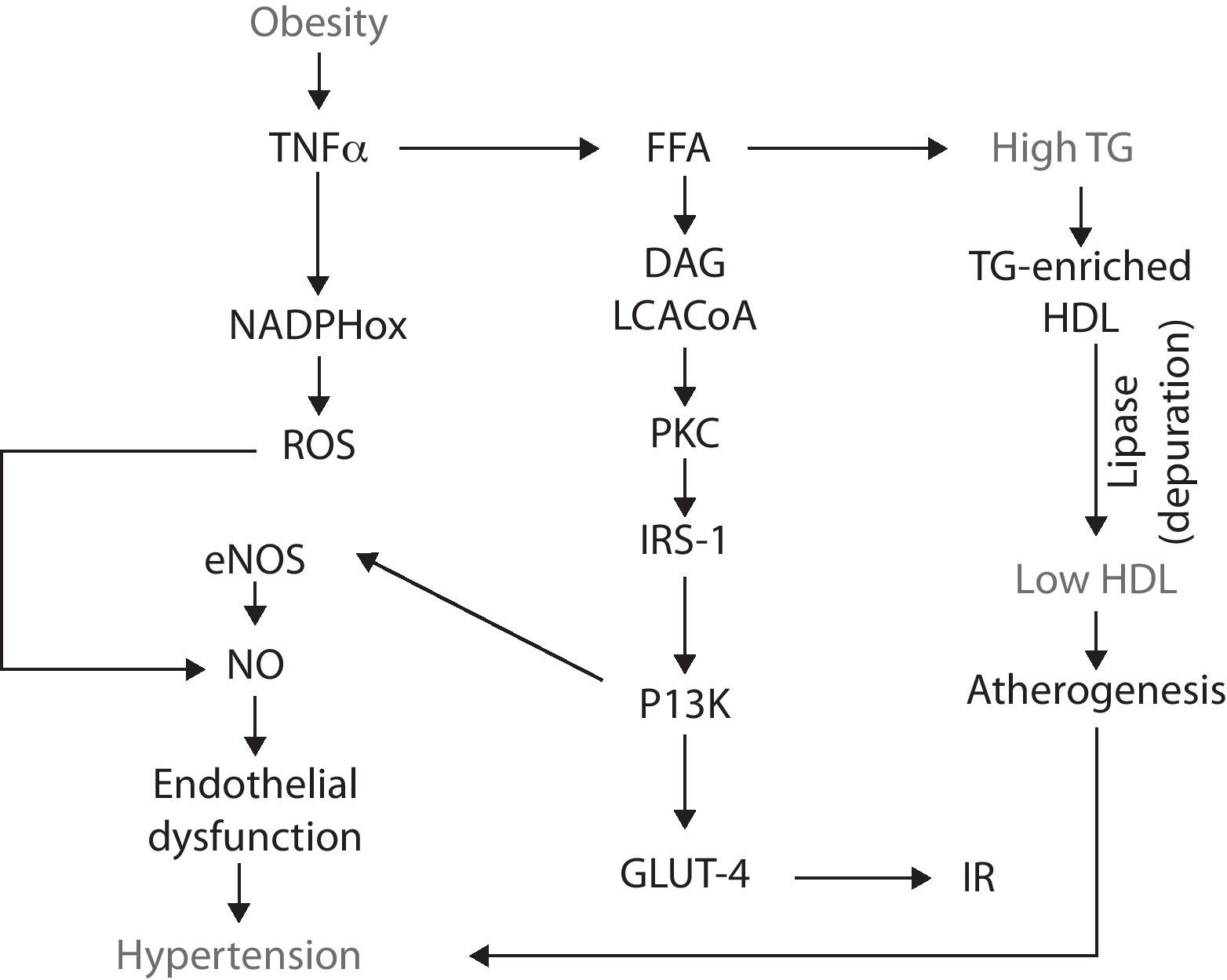
Obesity and the metabolic syndromeObesity is considered a pandemic disease with an estimated prevalence of almost 500 million adults; moreover, it is projected that by 2015 approximately 700 million – worldwide – will be obese.17,18 The importance of this disease relies on the fact that, by itself, obesity is a major risk factor for various diseases such as insulin resistance (IR) and type 2 diabetes, arterial hypertension and dyslipidemias (hypertrigliceridemia and low high-density lipoprotein levels).19 Such abnormalities, together, make up the metabolic syndrome (MS).20,21IR was initially proposed as the core of the MS.22 However, it is now recognized that obesity may be the triggering agent for each component of the syndrome since many of the underlying mechanisms correspond to the deleterious effects of certain adipokines secreted by the excess adipose tissue.23–25 In this regard, the excessive fat mass that defines obesity tends to be accumulated at the abdominal region. Peri-visceral fat, unlike the subcutaneous adipose tissue, is a metabolically active organ26,27 that releases several adipokines – which have been postulated as the link between obesity and the pathophysiology of MS – such as tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), transforming growth factor β (TGFβ), resistin, plasminogen activator inhibitor-1 (PAI-1), and leptin.28–33
It has been suggested that, in obesity, fat cells “oversynthesize” a chemokine, the monocyte chemoattractant protein-1 (MCP-1), that recruits circulating monocytes, which, now accumulated in the adipose tissue, differentiate into macrophages which produce high levels of TNF-α34–36 initiating a vicious circle from which the MS begins.
Obesity-IRTNF-α stimulates lipolysis, thus promotes the releasing of free fatty acids (FFA) into the circulation. The catabolism of FFA by skeletal muscle, increases intramyocellular levels of long chain acylCoA and diacylglycerol. These two molecules are powerful allosteric activators of protein-kinase C (PKC), which, in turn, phosphorylates the insulin receptor substrates-1 (IRS-1) in threonine and serine amino acid residues, which impairs the functional phosphorylation on tyrosine residues.37,38 Such misguided phosphorylation of IRS-1 prevents the activation of the insulin receptor, consequently disrupting the downstream signaling cascade and, in turn, impairs the glucose transporters (GLUT-4) from migrating and merging within the cellular membrane. As a result, hyperglycemia and IR develop.39,40
IR-endothelial dysfunction (hypertension)IR is defined as a condition in which insulin is unable to perform many of its roles, for example, those related to metabolism and cardiovascular function.41 At the endothelial level, insulin does not have a metabolic role, i.e., it is not related with the recruitment of glucose transporters. Insulin rather binds to specific receptors associated with endothelial function by activating the phosphatidylinositol 3-kinase (PI3K) pathway; this pathway is responsible for the phosphorilation and activation of endothelial nitric oxide synthase (eNOS). Thus insulin has a vasodilatory effect.42,43 However, when IR occurs, the PI3K pathway becomes affected, resulting in endothelial dysfunction due to a decrease in the bioavailability of nitric oxide (NO) and, consequently, a decrease in its capability to induce vasodilation. Endothelial dysfunction is a pathological state defined as an imbalance in the production of vasodilating and vasocontricting substances, thus the endothelium is not able to carry out its normal physiologic mechanisms.44-46
Obesity-oxidative stress-endothelial dysfunctionObesity is not only associated to endothelial dysfunction through the alterations in fat accumulation, TNF-α and IP3K, but also through other abnormalities such as oxidative stress. Oxidative stress is an imbalance between the production of reactive oxygen and nitrogen species (ROS/RNS) and the cell's ability to buffer them. Obesity-associated oxidative stress is enhanced by the increased releasing of adipokines which, in turn activate NADPH oxidase, resulting in the consequent increase in ROS (i.e., superoxide radical) production. In the endothelium, superoxide rapidly reacts with NO and forms peroxynitrite, an even more reactive specie. By this, ROS production decreases NO availability and, as a result, it contributes to endothelial dysfunction.47 NADPH oxidase is not the only mechanism by which obesity increases oxidative stress: obese subjects have an increased oxygen consumption because of a greater mechanical load and myocardial metabolism. One of the negative consequences of this feature is the production of ROS derived from the mitochondrial respiration and the loss of electrons along the transport chain, resulting in the formation of diverse reactive radicals such as superoxide.48
Dyslipidemias-endothelial dysfunctionEndothelial dysfunction can also develop as a consequence of another MS feature: atherogenic dyslipidemias.49 It has been established that obesity is an IR-prone entity, thus insulin fails to suppress the lipolysis already enhanced by TNF-α, which results in higher FFA plasmatic concentrations. These lipids enter the circulation and eventually reach the liver, where they serve as substrates for the synthesis of triglyceride (TG). As more FFA reach the hepatocyte, more TG are produced and, because they are released into the bloodstream, they eventually induce hypertriglyceridemia.50 Such excessive TG are coupled in the liver to high-density lipoproteins (HDL), resulting in a TG-enriched HDL. This molecule has high affinity for the hepatic lipase, thus the lipoprotein is rapidly depurated from the circulatory system, decreasing HDL to participate in the reverse transport of cholesterol, thus favoring atherogenesis and hypertension51,52 (Fig. 1).
New approaches to the understanding of MS: PPARs, SIRTs and PGCs
Recent findings have pointed out the participation of mitochondrial function in obesity and its comorbidities, including the MS. Mitochondria are organelles found in most of the human cells and more abundantly in cells with a high metabolic activity (e.g., myocytes, hepatocytes, neurons).53 Mitochondria have an important role in terms of energy homeostasis (ATP production and energy expenditure), substrate metabolism, disposal of ROS, adipogenesis and mature adipocyte function.54 The term mitochondrial dysfunction refers to the mitochondria inability to perform the previously mentioned roles.
On this regard, obesity has been associated with mitochondrial dysfunction. Several studies have reported a reduction in the size, morphology, number and activity of mitochondria,55,56 suggesting that the perturbation of the mitochondrion's function causes energy imbalances due to a defective network of interactions between certain regulator proteins such as peroxisome proliferator-activated receptors (PPARs), sirtuins (SIRTs), and PPAR coactivator 1 alpha (PGC-1α), among others.
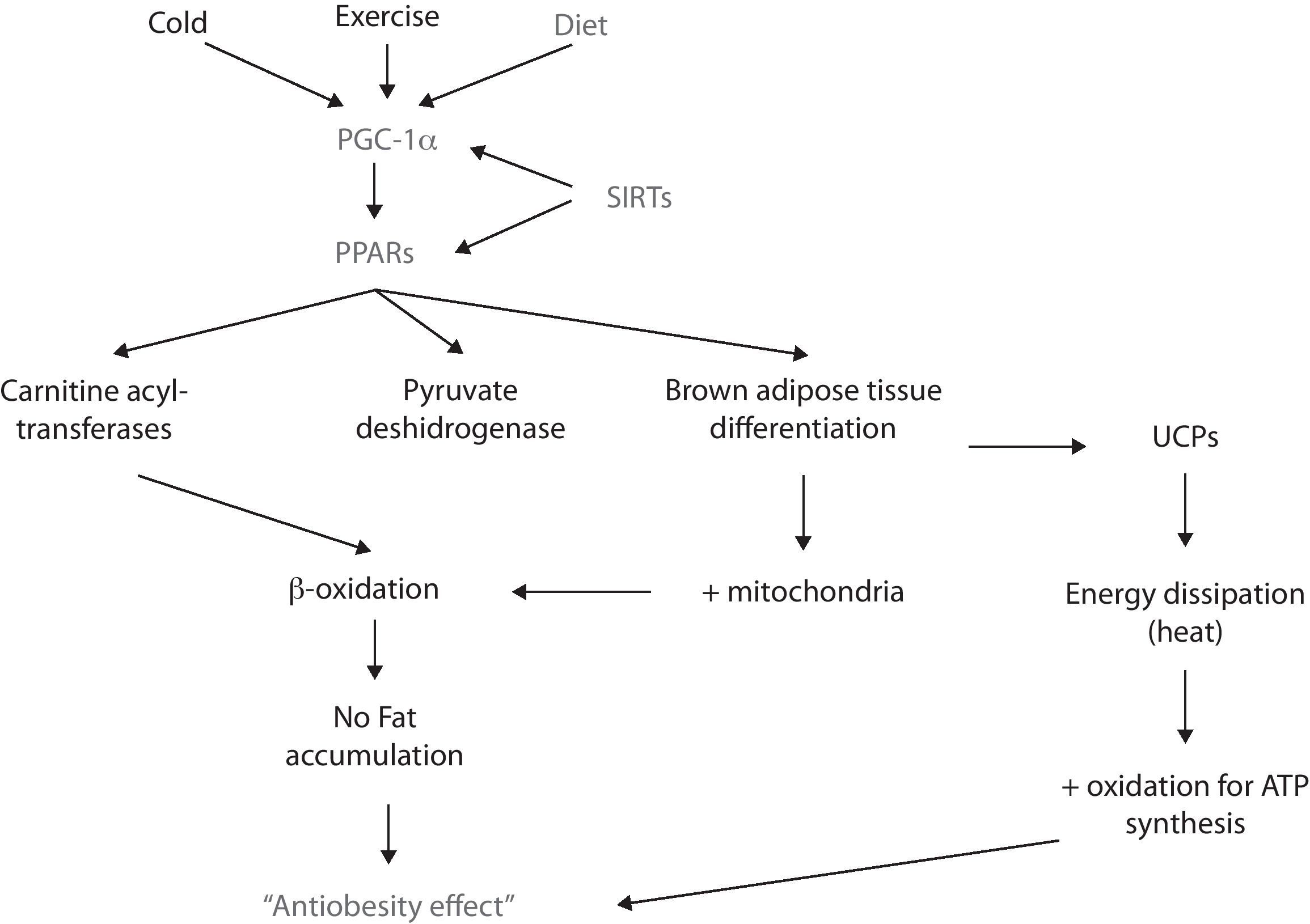
PPARRecently, there has been stated that a family of nuclear receptors called peroxisome proliferator-activated receptors (PPARs) alpha, beta-delta and gamma, are ligand-activated transcription factors that regulate the expression of certain genes by binding to the DNA and activating its transcription.57 In this context, PPARs participate in lipid catabolism and storage, adipocyte differentiation, prevention of weight gain, and suppression of pro-inflammatory cytokines, among other roles.58,59
PPARα and PPARβ/δ participate in lipid metabolism by promoting betaoxidation (i.e., fatty acid oxidation) in liver and skeletal muscle.60 Such increase in lipid catabolism is achieved since the activation of PPAR increases the synthesis of carnitine acyl-transferases and pyruvate deshydrogenase (both of them are rate limiting enzymes in the oxidation of fatty acids)61,62; hence, the oxidation pathway is favored and lipids become the principal source of metabolites for ATP synthesis which, consequently impairs them from being stored.
For its part, PPARγ participates in the adipogenesis of both white and brown adipose tissue. The latter has a higher oxidative capacity since it has more mitochondria; moreover, brown adipose tissue dissipates energy rather than storing it due to the uncoupling of the oxidative phosphorilation chain. Of high relevance in terms of obesity is that PPARγ’s activity can favor brown adipose tissue differentiation.63 In fact, recent in vitro studies have shown that exposing white adipose tissue cultures to PPARγ ligands (e.g., rosiglitazone) induces a “browning” of the white adipocytes, featuring an increase in the number of mitochondria, as well as an enhancement in its structure – i.e., mitochondrial cristae – and an increase of fatty acid oxidation.64,65 However, there is not conclusive evidence yet since the uncoupling feature in brown adipocytes is essentially conferred to the uncoupling proteins (UCP) depends on various receptors and transcription factors rather than just PPAR.
PPARs, then, are an interesting target for the treatment of obesity and MS, either by modulation of the inflammatory process and/or by activating macronutrient oxidation pathways (i.e., glucose and lipids).66-69 In fact, two drugs that were frequently included in MS treatment, thiazolidinediones and fibrates, are synthetic pharmacological ligands for PPARs.70,71 However, these drugs have also been associated with significant adverse effects including cholelithiasis, fluid retention, congestive heart failure, and venous thrombosis.72 Hence, there is a need to develop safer and novel agonists of PPAR as obesity and MS are becoming a pandemia.
PGC-1αAnother molecule implicated in energy balance and obesity is PPAR coactivator 1 alpha (PGC-1α). This coactivator interacts with transcriptional factors (such as PPARs, SIRTs, etc.) and, thus enhances the expression of genes involved in energy metabolism, thermogenesis, glucose and lipids metabolism, mitochondrial function and biogenesis, adipocyte differentiation and energy expenditure, such as: nuclear respiratory factor 1 (NRF1), PPARα and PPARγ, Farsenoid X receptor (FXR), glucocorticoid receptor (GR), among others.73,74
The expression of PGC-1α is regulated by different stimuli, for example, temperature. In this matter, the exposure to cold enhances PGC-1α synthesis via protein-kinase A (PKA).75 PGC-1α then coactivates PPARα and – this way – enhances the synthesis of certain proteins (e.g., UCP-1), that uncouple the respiratory chain and ATP synthesis, thus favors energy dissipation as heat in both brown adipose tissue and skeletal muscle.76,77 However, since in large mammals brown adipose tissue is not present in a significant amount, skeletal muscle is considered as the primary thermogenic and energy metabolizing tissue.
Another stimulus that promotes the expression – mainly in skeletal muscle – of PGC-1α is exercise. There are different types of skeletal muscle fibers: I, IIa, IIb and IIc. The first two have a higher oxidative metabolism since they contain more mitochondria. PGC-1α induces remodelation of skeletal muscle: from type IIb to type I and IIa; this change is corroborated by a redder muscle color and an increase in oxidation of macronutrients.78
New findings have shown that PGC-1α expression is downregulated in metabolic syndrome and obesity.79,80 Recently, it has been stated that high concentrations of FFA participate in the methylation of PGC-1α promoter, which inhibits its expression and leads to mitochondrial dysfunction81; since its metabolites – such as DAG and ceramides – induce so. It also impairs mitochondrial biogenesis, decreases the oxidation of lipids and thermogenesis. Hence, energy storage and the development of obesity and all of its metabolic alterations are favored. For these reasons, there's great interest in using PGC-1α as a powerful modulator of energy balance and a possible agent for treating obesity and the MS.
SirtuinsRecent studies have shown that another family of proteins is involved in the complex relationship between obesity and the MS: sirtuins (SIRTs). Seven isoforms (SIRT 1–7) have been found in mammals and they are expressed in the cytoplasm, nuclei and mitochondria. SIRTs activate metabolism-related proteins – such as PGC-1α – by deacetylating lysine residues.82,83
For example, SIRT-1 deacetylates PGC-1α and, by this means, regulates mitochondrial biogenesis. In skeletal muscle, such deacetylation increases mitochondrial activity, leading to higher energy expenditure by means of fatty acid oxidation, thermogenesis and endurance; these three effects prevent obesity.84,85
Being a NAD-dependent enzyme, SIRT activity is influenced by the NAD/NADH ratio: thereby, NAD increases SIRT activity, whereas NADH decreases it. This means that sirtuin activity is related to the level of available energy: NAD is in high concentrations when the molecular pathways of energy metabolism (glycolisis, Krebs cycle, etc.) are not active – or have low activity – causing SIRTs to increase their activity. In fact, studies in yeast have shown that energy restriction decreases NADH levels and, consequently, SIRTs activity increases.84 Moreover, in animal models, prolonged fasting increases NAD levels and, consequently SIRTs activity does as well.86
Although not all metabolic pathways in humans are completely understood, it is now accepted that SIRT-1 participates in obesity and MS in several ways; for example, by interacting with PPARγ.87 SIRT-1 represses PPARγ transcriptional activity, thus leading to the inhibition of adipogenesis (defined as the mechanism by which preadipocytes differenciate into mature adipocytes) and the activation of lipolysis in white adipose tissue.88 This – eventually – could lead to weight loss in terms of body fat and, thus prevents/reverses obesity (Fig. 2).
Perspectives on a new anti-obesity therapy
From the previous lines, we can argue the importance of preventing/reversing obesity. Weight loss reduces the risk of developing type 2 diabetes and cardiovascular disease, among other pathologies. In fact, losing weight has proven to decrease arterial pressure in hypertensive patients, reduce plasmatic TG levels and total cholesterol along with increasing HDL levels; finally, it has also shown to improve glycemia in patients with type 2 diabetes and IR.89
Nowadays, a wide range of anti-obesity treatments are available although they all have the same basic principle: leading to a negative energy balance (i.e., when energy expenditure is greater than energy intake, forcing the organism to utilize its reservoirs in order to meet the nutritional requirements).89
- •
Nutritional therapy. Refers to hypoenergetic and modified in nutrient composition diets. Weight loss is usually obtained by lessening in 500kcal the patient's current diet; sometimes, a high-protein and low-carbohydrate diet is prescribed, although this resource is not recommended for long-term goals.
- •
Pharmacologic therapy. Sibutramine and orlistat are the most studied/prescribed drugs. The first one is a serotonine reuptake inhibitor whose weight loss actions are attributed to appetite suppression (i.e., diminishes energy intake). Conversely, orlistat is the only lipase inhibitor approved for weight loss treatment; as its name implies, orlistat acts by binding to the active site of pancreatic lipase, preventing – this way – fat digestion and absorption (thus, it also diminishes energy intake since less fat enters the body).
- •
Surgical approaches. These often result in weight loss by restricting the size of the stomach (thus limiting the amount of food consumed) or by bypassing a portion of the intestine (which reduces food digestion and, consequently, nutrient absorption).
Even though modifications towards a “healthy lifestyle” (i.e., correct diet and sufficient physical activity) are still the cornerstone in the treatment of obesity and MS, these interventions have shown low efficacy in the long term: a low rate of therapeutical attachment to nutritional intervention, adverse effects to drugs or surgery, etc. On the other hand, there are numerous over-the-counter dietary supplements that claim to be beneficial in losing weight and/or improving the MS features. However, these drugs lack of scientific background and – the majority of the time – were not clinically, nor preclinically, tested before, making them a potential threat to an individual's health.90
Since obesity has an increasing prevalence, it's not a surprise that the scientific community is interested in the development of newer agents that induce/activate all of the previously described molecules (SIRT, PPAR, PGC-1α) and, hence, can be employed in the treatment of obesity and MS. Some of this expected drugs are currently available; for example, thiazolidinediones bind to PPAR and, hence, induce brown adipose tissue differentiation, mitochondrial biogenesis and lipid oxidation. Unfortunately, they – as other pharmacologic agents do – also exhibit potentially significant adverse effects.
Fortunately, research has turned its attention to a sometimes underestimated agent: food. Several components of food have shown to perform regulatory functions related to energy balance and, thus to obesity. This means that obesity can be prevented and/or treated – in terms of nutrition – not only by a caloric/energetic restriction but also by the participation of specific active substances present within diet that interact with the patient's genetic material and induces the expression of certain genes and the synthesis or activation of proteins that potentially benefit the so called obesometabolic dysfunction. In fact, there's a whole novel field called nutrigenomics, which refers to the study of the influence of an individual's diet – and some specific food components – on the genome. This “new” field emerged after scientists recognized that nutrients – as drugs do – have the ability to interact and modulate physiological and pathophysiological molecular mechanisms.91 Among such nutrients, flavonoides exhibit a particularly interesting potential.
Flavonoids are a wide group of polyphenolic compounds that are present in almost every plant and vegetable. Some of them have already shown benefits in terms of obesity and MS. For example, recent investigations demonstrated that resveratrol (a flavonoid contained in high quantities in grapes and red wine) increases SIRT-1 activity and – this way – improves mitochondrial function, probably through allosteric interaction and the activation of PGC-1α in skeletal muscle.56,92 Moreover, in rodent models of obesity, resveratrol has been claimed to be not only an activator of SIRT-1 but an actual inductor of its gene.93–95 These mechanisms are manifested as improvements in endothelial function, decreases in lipid deposition and fat storage, muscular fiber switch, increased mitochondria size, biogenesis and density, and an increase the oxidative capacity through fatty acid oxidation.92,96
Other nutrigenomic agents are licorice flavonoids. These have shown to reduce (almost inhibit) weight gain in mouse diet-induced obesity models. Moreover, these flavonoids supplementation impaired abdominal fat accumulation, evidenced not only by a lower weight of the mesenteric and perirenal adipose tissues, but also by smaller size adipocytes (approximately one-third to one-half the size of those of the control group). Licorice flavonoids also had an effect on the liver: upon histological examination, liquid droplets were abundant in the control group, but not in the supplemented group.99,100
A final example refers to polyunsaturated fatty acids (PUFAs). These have a particularly high affinity for PPARs, thus a diet high in PUFAs should translate in an increase in their actions including antiglycemic, antilipemic and anti-inflammatory effects.96 Isoflavones are another kind of polyphenols that exert effects through the PPAR pathway: for example, genistein not only binds directly and activates PPARα (lipogenic, as mentioned earlier) but it also is capable of downregulating PPARγ (adipogenic).97 Other food components that have shown PPAR affinity include capsaicin, ginger and various flavonoids such as naringenin. The latter is derived from orange and grapefruit and is has been reported to increase fatty acid oxidation through PPARa-mediated transcription.98
Concluding remarksAlthough studies performed so far are limited, research on the role that food exerts in the modulation of several molecular pathways has given an insight on its potential use as an alternative/complement to currently employed drugs.101 As more molecules are identified to be key regulators in energy balance – hence, in the development of obesity – and the field of nutrigenomics grows, it can be expected that nutritional management of obesity and the MS will provide therapeutic interventions specifically targeted to modulate the genetic expression of such molecules, thus improving mitochondrial function and increasing the efficiency to prevent and manage such diseases.
Conflict of interest statementThe authors declare they have no financial or personal situation that could give for conflict of interest in relation to the paper presented here.
ContributionAll authors contributed to the idea, data collection, analysis information, drafting, critical revision and final approval thereof.