




El consumo excesivo de alimentos hipercalóricos y de alto contenido en grasas saturadas produce una dislipidemia aterogénica. En este estudio hemos analizado los efectos del activador de PPARβ/δ GW501516 sobre la hipertrigliceridemia inducida por una dieta rica en grasas.
MetodologíaRatones macho fueron distribuidos aleatoriamente en 3grupos: control (dieta estándar), dieta grasa (high fat diet [HFD], 35% grasa en peso, 58% kcal procedentes de grasa) y dieta grasa más GW501516 (3mg/kg/día). La duración del tratamiento fue de 3semanas.
ResultadosLa HFD causó hipertrigliceridemia acompañada de una reducción de los niveles hepáticos de la proteína AMPK fosforilada y de los niveles de ARNm Pgc-1α y lipina1. Estos efectos fueron revertidos por el tratamiento con GW501516. El mantenimiento de la AMPK fosforilada tras el tratamiento con GW501516 podría deberse al aumento de la relación AMP/ATP. GW501516 incrementó los niveles de proteína lipina1 nuclear acompañado por una amplificación de la vía PGC-1α-PPARα y un aumento de la actividad de unión al ADN de PPARα, así como el incremento en la expresión de los genes diana de PPARα implicados en la β-oxidación de ácidos grasos. GW501516 también aumentó los niveles plasmáticos de β-hidroxibutirato, producto final de la β-oxidación hepática. Finalmente, GW501516 incrementó los niveles del ligando endógeno de PPARα, 16:0/18:1-fosfatidilcolina, y aumentó la expresión del receptor de las VLDL en hígado.
ConclusiónEl efecto hipotrigliceridemiante de GW501516 en ratones sometidos a HFD se acompaña de un aumento de los niveles de la AMPK fosforilada y de un aumento de la vía PGC-1α-lipina1-PPARα.
Excessive consume of hypercaloric and high in saturated fat food causes an atherogenic dyslipidemia. In this study we analyzed the effects of PPARβ/δ activator GW501516 on the hypertriglyceridemia induced by a high-fat diet.
MethodsMale mice were randomized in three groups: control (standard chow), high fat diet (HFD, 35% fat by weight, 58% Kcal from fat) and high fat diet plus GW501516 (3mg/Kg/day). Treatment duration was three weeks.
ResultsHFD-induced hypertriglyceridemia was accompanied by a reduction in hepatic levels of phospho-AMPK and in PGC-1α and Lipin1 mRNA levels. All these effects were reversed by GW501516 treatment. The lack of changes in phospho-AMPK levels after GW501516 treatment in HFD-fed animals could be the result of an increase in the AMP/ATP ratio. GW501516 treatment also increased Lipin1 protein levels in the nucleus, led to the amplification of the PGC-1α-PPARα pathway and increased PPARα DNA-binding activity, as well as the expression of PPARα-target genes involved in fatty acid oxidation. GW501516 also increased β-hydroxibutirate plasmatic levels, a hepatic β-oxidation end product. Finally, GW501516 increased the hepatic levels of the PPARα endogenous ligand 16:0/18:1-PC and the expression of the VLDL receptor.
ConclusionThese data indicate that the hypotriglyceridemic effect of GW501516 in mice subjected to HFD-fed mice is accompanied by an increase in phospho-AMPK levels and the amplification of the PGC-1α-Lipin1-PPARα pathway.
La ingesta excesiva de calorías y la gran disponibilidad de nutrientes han permitido una continua progresión de la incidencia de la obesidad y la resistencia a la insulina, principales factores responsables del desarrollo del síndrome metabólico. Este síndrome se caracteriza por la presencia de una dislipidemia que se inicia con la sobreproducción hepática de lipoproteínas de muy baja densidad (VLDL) que transportan triglicéridos, seguida de la aparición de partículas LDL más pequeñas y densas y de una reducción en los niveles de colesterol HDL e hiperlipidemia postprandial1,2. Todas estas alteraciones convierten la dislipidemia aterogénica en un factor de riesgo muy importante en el desarrollo de la enfermedad aterosclerótica3, razón por la cual es muy importante buscar nuevos tratamientos para reducir los niveles de triglicéridos plasmáticos.
Entre los nuevos tratamientos farmacológicos para prevenir el aumento de los niveles de triglicéridos, los ligandos de los receptores activados por proliferadores peroxisómicos (peroxisome proliferator-activator receptor [PPAR]) β/δ han despertado un gran interés en los últimos años. Los PPAR son miembros de la superfamilia de los receptores nucleares hormonales que regulan la transcripción de genes implicados en diversos procesos biológicos4. La familia de los PPAR está formada por 3subtipos codificados por genes independientes, PPARα (NR1C1, según el sistema unificado de nomenclatura por la superfamilia de los receptores nucleares), PPARβ/δ (NR1C2) y PPARγ (NR1C3)5. De los 3subtipos, PPARα fue el primero en ser identificado y es la diana molecular de los fibratos. Este subtipo se expresa principalmente en tejidos con alta capacidad catabólica como el hígado, el tejido adiposo marrón, el riñón, el corazón y el músculo esquelético6. El PPARγ tiene un patrón de distribución más restringido, expresándose principalmente en el tejido adiposo marrón y blanco, en el colon y los macrófagos, mientras que su expresión es limitada en el músculo esquelético y el corazón. Por otro lado, el PPARβ/δ se expresa de manera ubicua, incluyendo tejidos altamente metabólicos como el hígado, el músculo y el tejido adiposo, y su papel en el síndrome metabólico ha sido estudiado en los últimos años7-9. Entre otros efectos, el tratamiento con el ligando de PPARβ/δ de alta afinidad GW501516 ha demostrado disminuir los niveles de triglicéridos plasmáticos10. En estudios realizados en ratones deficientes en PPARβ/δ el efecto hipotrigliceridemiante de este subtipo de receptor nuclear se ha asociado con cambios en la producción hepática y la eliminación de las VLDL11, pero se desconoce si otros mecanismos podrían estar implicados. Es interesante destacar que el principal factor que determina la secreción de triglicéridos hepática es la disponibilidad de ácidos grasos12. En el hígado, los ácidos grasos pueden ser incorporados en los triglicéridos u oxidados en el proceso de β-oxidación mitocondrial. Así, un aumento en la oxidación de los ácidos grasos en el hígado podría reducir la disponibilidad de estos ácidos grasos y, en consecuencia, reducir la secreción de triglicéridos hepáticos. Sin embargo, se desconoce si el efecto hipotrigliceridemiante causado por la activación del PPARβ/δ supone un aumento de la β-oxidación hepática y los mecanismos implicados.
El paso limitante de la β-oxidación mitocondrial es el transporte de los ácidos grasos al interior de la mitocondria a través de la carnitina palmitoiltransferasa-1 (CPT1a). Este transportador de ácidos grasos se encuentra bajo control tanto de los PPAR como de la cinasa activada por adenosinmonofosfato (AMP) (AMP-activated protein kinase [AMPK]), que detecta niveles bajos de adenosintrifosfato (ATP) y, en respuesta, aumenta el metabolismo oxidativo13 reduciendo los niveles de malonil-CoA. Además, hay que destacar que la activación de PPARβ/δ puede aumentar la actividad de la AMPK y que el tratamiento en células de músculo esquelético humanas con GW501516 puede aumentar la oxidación de los ácidos grasos a través de PPARβ/δ y la AMPK14.
Recientemente se ha descrito el papel de una nueva proteína, la lipina1, que determina si los ácidos grasos son incorporados a los triglicéridos o son conducidos hacia la β-oxidación mitocondrial. La expresión y la localización de la lipina1 controlan la secreción de los triglicéridos hepáticos15. Así, en el citoplasma, la lipina1 promueve la acumulación de triglicéridos y la síntesis de fosfolípidos actuando como una fosfatasa del ácido fosfatídico dependiente de Mg2+ (phosphatidic acid phosphatase-1 [PAP1]). Por otro lado, cuando la lipina1 se localiza en el núcleo actúa como un co-activador transcripcional ligado a la oxidación de los ácidos grasos que regula la inducción de genes diana del complejo PGC1α-PPARα16. La lipina1 induce la expresión de genes diana de PPARα y forma un complejo junto con PPARα y PGC1α, permitiendo la inducción de genes diana implicados en la oxidación de ácidos grasos, tales como Cpt1a y la deshidrogenasa de acil-CoA de cadena media (medium chain acyl-coA dehydrogenase [Mcad])16.
El objetivo de este estudio ha sido evaluar los efectos del activador de PPARβ/δ GW501516 sobre la hipertrigliceridemia inducida por una dieta rica en grasa (high fat diet [HFD]) y sus efectos sobre la β-oxidación hepática.
MétodosEl ligando de PPARβ/δ GW501516 se obtuvo de Alexis Biochemicals (Lausen, Suiza). [γ-32P]dATP (3000Ci/mmol) fue proporcionado por Amersham Biosciences (Pisacataway, NJ, EE.UU.). El resto de productos químicos se obtuvieron de Sigma-Aldrich.
AnimalesRatones de 5 semanas de edad se mantuvieron bajo condiciones estándar de iluminación (ciclos de 12h luz/oscuridad) y de temperatura (21±1°C) y se alimentaron con dieta estándar (Harlan, Barcelona, España) durante 5días antes del inicio del estudio. Los animales se distribuyeron aleatoriamente en 3grupos experimentales (n=12 cada uno): uno alimentado con dieta estándar, otro alimentado con dieta rica en grasas tipo Western (HFD, 35% grasa por peso, 58% kcal en grasa, Harlan Ibérica S.A., Barcelona, España) más una dosis diaria por sonda oral de vehículo (0,5% w/v carboximetilcelulosa de viscosidad media), y animales alimentados con HFD más una dosis diaria por sonda oral de 3mg/kg/día del agonista de PPARβ/δ GW501516 disuelto en el vehículo. Antes del término del tratamiento se realizó un test de tolerancia a la glucosa a los ratones en ayunas durante 4h. Los animales recibieron 2g/kg por peso corporal de glucosa vía intraperitoneal y se recolectaron muestras de sangre a los 0, 20, 40 y 90min. El peso corporal de los ratones y el consumo de dieta fue comprobado regularmente durante el período de tratamiento. Tras 3semanas de tratamiento los animales fueron sacrificados utilizando isoflurano como anestesia. Tras la recolección de sangre, se analizaron los niveles plasmáticos de triglicéridos, glucosa (Bayer Iberia, Sant Joan Despí, España), colesterol, ácidos grasos libres (Wako, Japón), insulina (Amersham), adiponectina y leptina (Linco, St. Charles, MO, EE.UU.). Las muestras de hígado se congelaron en nitrógeno líquido y fueron guardadas a –80°C. Estos experimentos se realizaron conforme a la Guía para el Cuidado y Uso de Animales de Laboratorio publicada por el Instituto Nacional de Salud de los Estados Unidos (NIH Publication No. 85-23, revisada en 1996). Todos los procedimientos fueron aprobados por la Comisión de Bioética de la Universidad de Barcelona, como indica la Ley 5/21 de julio de 1995 aprobada por la Generalitat de Cataluña.
Análisis de los niveles de ARNmLos niveles de ARNm se estudiaron mediante la reacción en cadena de la polimerasa de transcripción reversa (RT-PCR) tal como se ha descrito previamente17. El ARN total se aisló utilizando el reactivo Ultraspec (Biotecx, Houston, EE.UU.). El ARN total aislado mediante este método no está degradado y está libre de contaminaciones por ADN o proteínas. Las secuencias de los primers sentido y antisentido utilizados para la amplificación fueron: Lipina1, 5’-CTGCAGACAGGTTGACGCCAA-3’ y 5’-TCTGGTGGATGAGCAGTCCCC-3’; Pgc-1α, 5’-CCCGTGGATGAAGACGGATTG-3’ y 5’-GTGGGTGTGGTTTGCTGCATG-3’; Pparα, 5’-GGCTCGGAGGGCTCTGTCATC-3’ y 5’-ACATGCACTGGCAGCAGTGGA-3’; Cpt1a, 5’-TATGTGAGGATGCTGCTT-3’ y 5’-CTCGGAGAGCTAAGCTTG-3’, Mcad, 5’-TGGAAAGCGGCTCACAAGCAG-3’ y 5’-CACCGCAGCTTTCCGGAATGT-3’; Cte (cytosolic thioesterase) 5’-CAGCCACCCCGAGGTAAAAGG-3’ y 5’-CCTTGAGGCCATCCTTGGTCA-3’; Cept1(cholineethanolamine phosphotransferase) 5’-GCTAGGTGAGCCGCTCAGTGC-3’ y 5’-ATGGTGCCTCCTCCGTGACTG-3’; receptor de las Vldl (Vldl-r) 5’-GTTCAAGTGCAGAAGCGGGGA-3’ y 5’-CCGGGTTTTGGCATTCATCAA-3’; y Aprt (adenosyl phosphoribosyl transferase), 5’-AGCTTCCCGGACTTCCCCATC-3’ y 5’-GACCACTTTCTGCCCCGGTTC-3’. La amplificación de cada gen reveló una única banda de tamaño esperado: (lipina1: 225pb, Pgc-1α: 228pb, Pparα: 654pb, Cpt-1a: 629pb, Mcad: 216pb, Cte: 244pb, Cept1: 233pb, Vldl-r: 227pb, y Aprt: 329pb). Se llevaron a cabo experimentos preliminares para cada uno de los genes estudiados con varias cantidades de ADNc para determinar las condiciones no saturantes de la amplificación por PCR. Entonces, bajo estas condiciones, se evaluó la cuantificación relativa del ARNm por el método RT-PCR utilizado en este estudio18. Las bandas radiactivas se cuantificaron por escáner vídeo-densitométrico (Vilbert Lourmat Imaging). Los resultados para la expresión de los ARNm específicos siempre se presentan en relación con la expresión del gen control (Aprt).
Aislamiento de extractos nuclearesLos extractos nucleares se obtuvieron como se ha descrito previamente17.
Ensayo de retardación de la movilidad electroforéticaEl ensayo de retardación de la movilidad electroforética (electrophoretic mobility shift assay [EMSA]) se realizó usando una doble cadena de oligonucleótidos para la unión consenso de PPAR (sonda PPRE; 5’CAAAACTAGGTCAAAGGTCA-3’, Sta. Cruz Biotechnology, Santa Cruz, CA, EE.UU.), como se ha descrito previamente19.
Western-blotPara la obtención de proteína total el hígado de los ratones fue homogeneizado en tampón de lisis frío (Tris-HCl 5mM pH 7,4, EDTA 1mM, fenilmetilsulfonil floruro 0,1mM, ortovanadato sódico 1mM, aprotinina 5,4μg/ml). El homogeneizado fue centrifugado a 16,700×g durante 30min a 4°C.La concentración de proteína fue medida por el método Bradford. Los extractos proteicos se separaron por SDS-PAGE en geles de 10% y se transfirieron a una membrana immobilon-poliviniliden-dioflurada (Millipore, Bedford, MA, EE.UU.). Los análisis de western-blot se realizaron usando los anticuerpos contra AMPK, AMPK fosforilada Th172, ERK1/2, ERK1/2 fosforilada (Cell Signaling, Danvers, MA, EE.UU.), lipina1, PPARα, laminaB (Santa Cruz Biotechnologies) y β-actina (Sigma). La detección de estos anticuerpos se realizó con el kit de quimioluminiscencia EZ-ECL (Amersham). El tamaño de las proteínas detectadas se estimó basándose en marcadores estándar de peso molecular (Invitrogen, Barcelona, España).
Análisis de 1-palmitoil-2-oleil-fosfatidilcolina (16:0/18; 1-PC)Los lípidos totales de los homogeneizados de hígado se extrajeron por el método Bligh & Dyer20, se evaporaron y se disolvieron en metanol/agua (9:1). La separación total de los lípidos, la identificación y la cuantificación se llevaron a cabo por cromatografía líquida asociada a espectrometría de masas usando una bomba binaria Hitachi LaChrom Elite L-2130 y un inyector automático Hitachi L-2200 (Merck) acoplado con un espectrómetro de masas Bruker Esquire6000 con trampa de iones21,22. El efluente se dividió introduciendo 0,2ml/min en el interfaz de eletropulverización del espectrómetro de masas. El nebulizador se estableció a 30psi, el gas de secado a 8l/min y la temperatura de secado a 350°C. La columna usada fue una Supelcosil LC-18 de 5μm de tamaño de partícula, 250×2,1mm (Sigma-Aldrich) protegida con un cartucho Supelguard LC-18 de 20×2,1mm (Sigma-Aldrich). La fase móvil usada fue un gradiente del solventeA (metanol/agua/hexano/hidróxido de amonio, 87,5:10,5:1,5:0,5 v/v/v/v), del solventeB (metanol/hexano/hidróxido de amonio, 87,5:12:0,5 v/v/v) y del solventeC (metanol/agua, 9:1 v/v). El gradiente empezó al 100% deA, disminuyó linealmente al 50% deA (50%B) en 17,5min, y al 0%A (100%B) en 12,5min, mantenido al 100% deB durante 5min y entonces modificado a 100% deC en 3min, mantenido 9min y modificado a 100%B en 3min. La velocidad de flujo fue 0,5ml/min y el volumen de inyección, 80μl. La adquisición de datos se llevó a cabo en el modo de exploración completa y positiva, detectando las especies de PC como iones [M+H]+ con la corriente del capilar a –4000V. La fosfatidilcolina (PC) (16:0/18:1) fue caracterizada por espectroscopia de masas en tándem en monitorización de reacción múltiple y en modo negativo, con una adición post-columna de ácido acético para la formación del aducto [M+CH3CO2] (100μl/h). Como estándar interno para la curva de calibración y para la cuantificación se utilizó 1,2-dinonadecanoil-sn-glycero-3-fosfocolina (m/z=818,6).
Determinación de adenosintrifosfato, adenosindifosfato y adenosinmonofosfato por cromatografía líquida de alta eficaciaLos nucleótidos de adenina fueron separados por cromatografía líquida de alta eficacia usando una columna X-Bridge con un diámetro exterior de 3,5μm (100×4,6cm). La elución fue realizada con dihidrógeno fosfato de potasio 0,1mM, pH6, conteniendo hidrógeno sulfato de tetrabutilamonio 4mM y el 15% (v/v) de metanol. Las condiciones fueron las siguientes: 20μl de inyección de muestra, columna a temperatura ambiente, velocidad de flujo a 0,6ml/min y UV a 260nm.
Análisis estadísticoLos datos se presentan como media±desviación estándar de 5experimentos separados. Las diferencias significativas se establecieron por ANOVA de una vía, utilizando el programa GraphPad Instat (Software GraphPad V2.03) (GraphPad Software, CA, EE.UU.). Cuando se encontraron variaciones significativas se aplicó el test de comparación múltiple de Tukey-Kramer. Las diferencias se consideraron significativas con un valor de p<0,05.
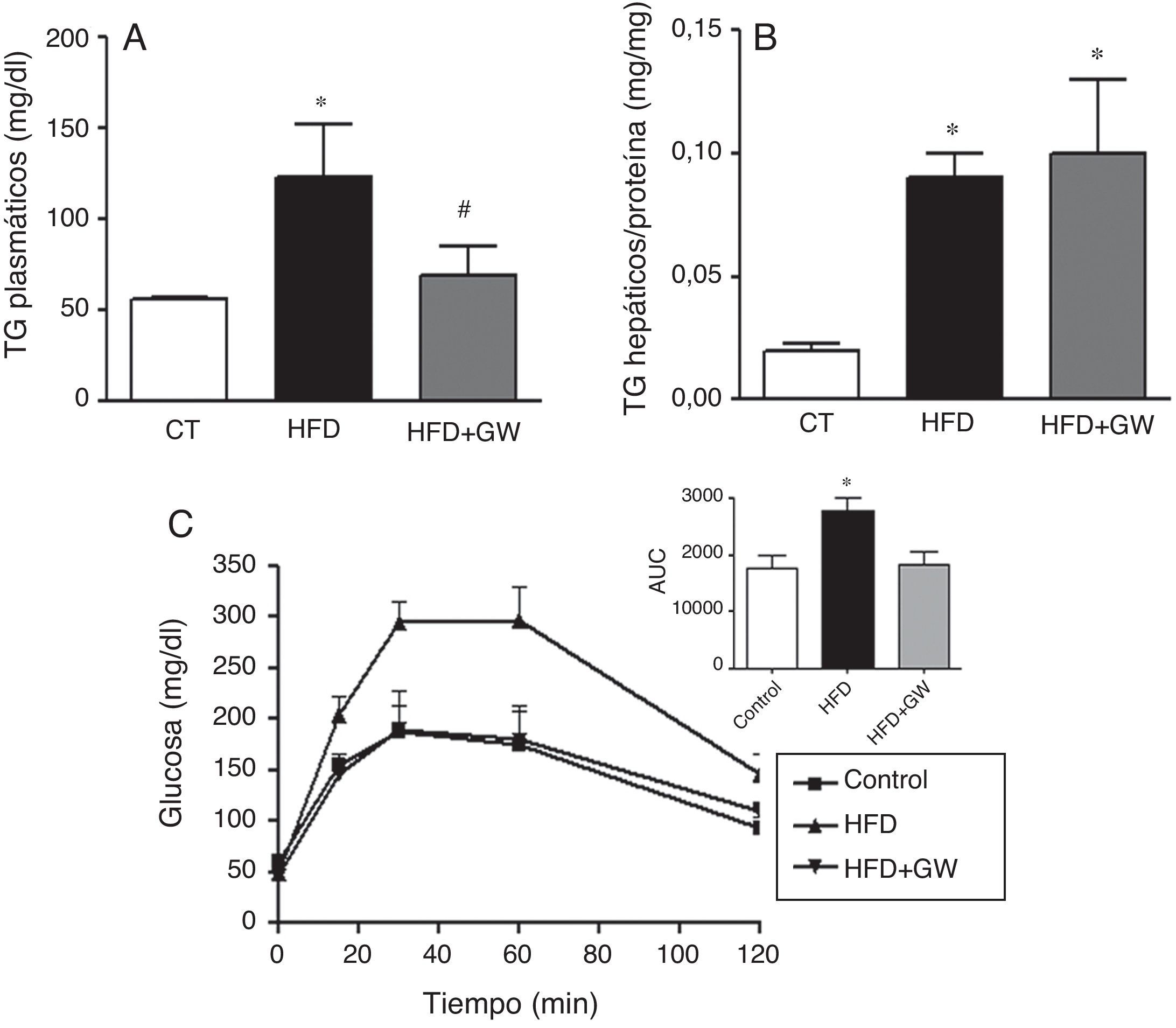
ResultadosEfectos del tratamiento con GW501516 sobre los niveles de triglicéridos plasmáticos, el contenido de triglicéridos hepático y el test de tolerancia a la glucosa en ratones alimentados con la dieta rica en grasaLa mayoría de estudios realizados con agonistas de PPARβ/δ están basados en tratamientos a largo plazo, permitiendo una importante reducción de peso y de grasa23,24, efectos que repercuten sobre el metabolismo lipídico y la sensibilidad a la insulina. Para prevenir la interferencia que provoca la pérdida de peso en los parámetros analizados, en este estudio los ratones fueron tratados con el agonista de PPARβ/δ durante 3semanas. Tras este período, los ratones que recibieron la HFD o la HFD más el tratamiento con GW501516 no mostraron diferencias significativas en el peso corporal en comparación con los ratones control (control: 30,4±2,8g, HFD: 30,0±2,5g, y HFD+GW501516: 31,4±2,7g), y el consumo de comida fue similar en todos los grupos (resultados no mostrados). Además, los ratones alimentados con la HFD o la HFD más GW501516 no mostraron cambios significativos entre los niveles plasmáticos de colesterol total, glucosa, insulina, adiponectina, leptina o ácidos grasos no esterificados comparados con los ratones control (resultados no mostrados). Por el contrario, los ratones alimentados con HFD mostraron un aumento de los niveles de triglicéridos comparados con los animales control (55±2,5 vs. 123±29mg/dl, inducción de 2,2veces, p<0,05), mientras que este aumento fue evitado por el tratamiento con GW501516 (69±16mg/dl, p<0,01 vs. ratones HFD) (fig. 1A). La HFD también aumentó el contenido de triglicéridos en el hígado (inducción de 4,5 veces, p<0,05) (fig. 1B). Por otro lado, el tratamiento con el fármaco no afectó este parámetro, coincidiendo con un estudio previo9. Al realizar el test de tolerancia a la glucosa, que evalúa la capacidad de organismo de ajustar los niveles de glucosa tras una inyección aguda de glucosa, los niveles de glucosa de los animales control mostraron un pico a los 30min y volvieron a niveles basales (fig. 1C). Como era esperado, los ratones alimentados con la HFD mostraron intolerancia a la glucosa, tal como se demuestra por el aumento significativo en el área bajo la curva (AUC). Por el contrario, los ratones alimentados con la HFD y tratados con GW501516 mostraron una mejor respuesta al cambio de la glucosa, y el AUC del test de tolerancia a la glucosa fue similar al de los ratones control.
 durante 3semanas. A) Niveles de triglicéridos plasmáticos. B) Contenido de triglicéridos hepáticos. C) Test de tolerancia a la glucosa y área bajo la curva (AUC). Los resultados se expresan como la media±DE (n=5). *p<0,05 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 previene la hipertrigliceridemia y la intolerancia a la glucosa en ratones alimentados con la HFD. Los ratones fueron alimentados con una dieta estándar o con la HFD con o sin GW501516 (3mg/kg/día) durante 3semanas. A) Niveles de triglicéridos plasmáticos. B) Contenido de triglicéridos hepáticos. C) Test de tolerancia a la glucosa y área bajo la curva (AUC). Los resultados se expresan como la media±DE (n=5). *p<0,05 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.
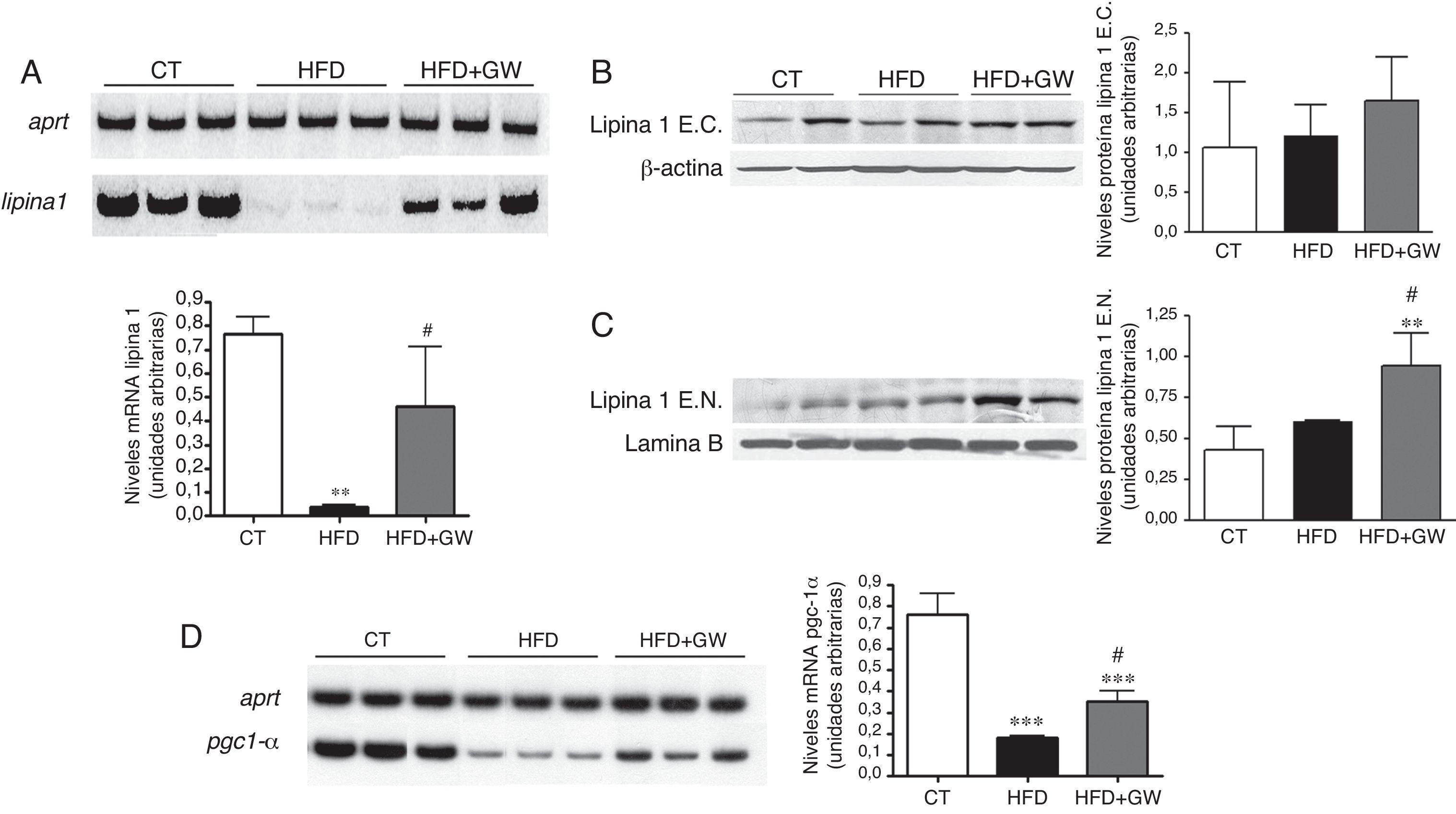
Para entender los efectos de la activación de PPARβ/δ sobre los ácidos grasos, si eran incorporados a los triglicéridos o por el contrario eran oxidados, se analizaron los niveles de lipina1, enzima que se encuentra bajo el control de la AMPK25. La HFD disminuyó notablemente los niveles hepáticos de expresión génica de la lipina1 en comparación con los ratones alimentados con la dieta estándar, reducción que no se observó en los ratones tratados con el activador de PPARβ/δ (fig. 2A). Los niveles proteicos de lipina1 citosólicos no se modificaron de manera significativa por la HFD o por el tratamiento con el fármaco (fig. 2B). Por el contrario, GW501516 aumentó los niveles proteicos de lipina1 en el núcleo (fig. 2C), donde actúa como co-activador transcripcional ligado a la oxidación de los ácidos grasos16. Además, la expresión hepática del co-activador Pgc-1α, implicado en la β-oxidación, disminuyó tras la alimentación con la HFD, pero esta reducción no se observó tras el tratamiento con GW501516 (fig. 2D).
 Niveles de ARNm de lipina1. Autorradiografía representativa y cuantificación normalizada respecto a los niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5). Niveles proteicos de lipina1 citosólica (B) y nuclear (C) en el hígado. Como control de carga se utilizó β-actina (extractos citosólicos) y laminaB (extractos nucleares). D) Niveles de ARNm de Pgc-1α. *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05; ##p<0,01 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 aumenta los niveles de lipina1 nuclear en el hígado de ratones alimentados con la HFD. A) Niveles de ARNm de lipina1. Autorradiografía representativa y cuantificación normalizada respecto a los niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5). Niveles proteicos de lipina1 citosólica (B) y nuclear (C) en el hígado. Como control de carga se utilizó β-actina (extractos citosólicos) y laminaB (extractos nucleares). D) Niveles de ARNm de Pgc-1α. *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05; ##p<0,01 vs ratones alimentados con la HFD.
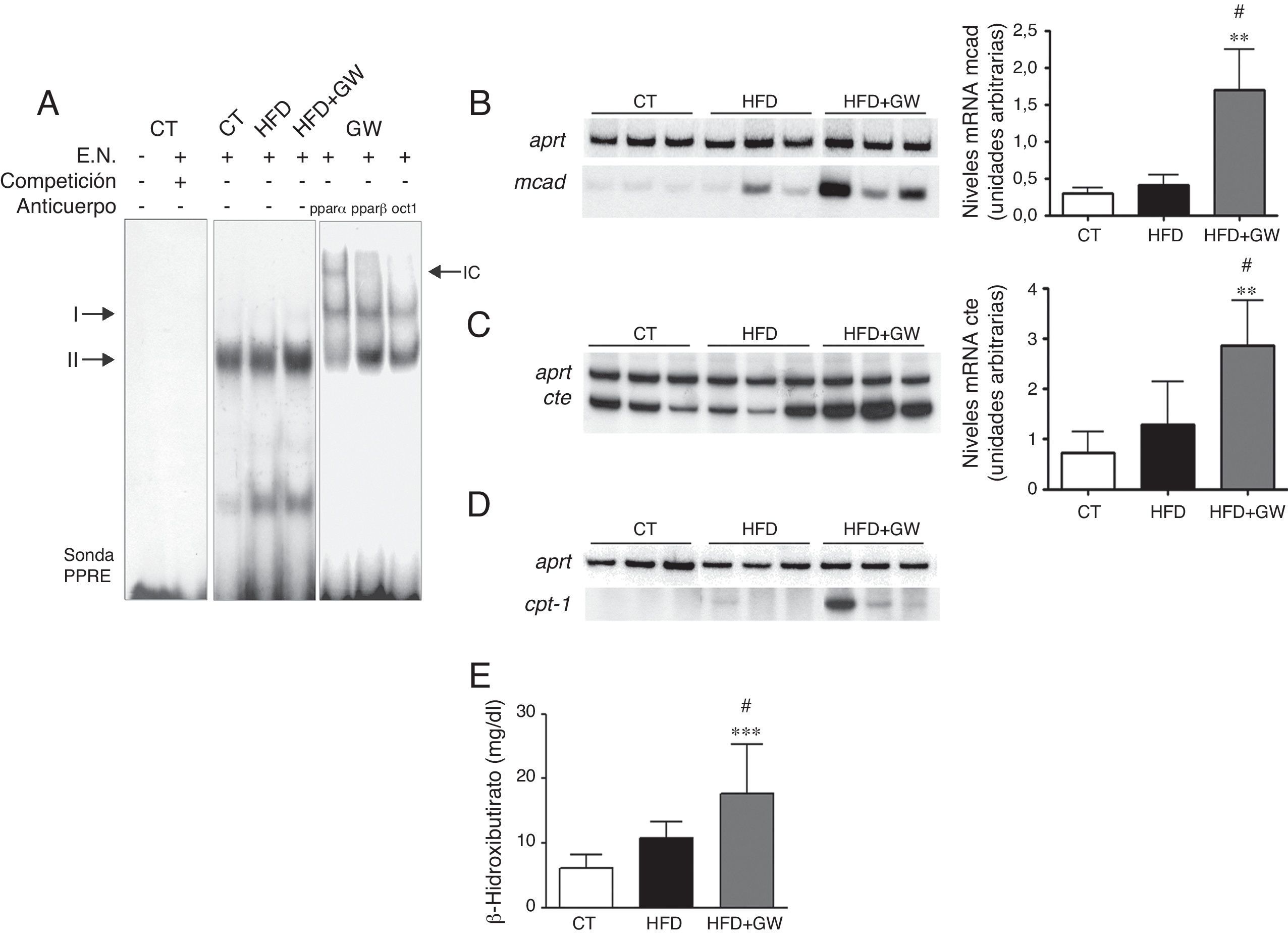
Se ha descrito que la sobreexpresión de lipina1 puede aumentar la expresión génica de Pparα y de sus genes diana como Cpt1a y Mcad, por lo que estudiamos si el aumento de lipina1 nuclear causado por GW501516 era responsable de estos cambios. Así, GW501516 aumentó los niveles génicos y de proteína nuclear de PPARα (resultados no mostrados). Además, dado que se ha demostrado que la lipina1 activa la transcripción de PPARα en cooperación con PGC-1α16, realizamos un EMSA para examinar el efecto del tratamiento con GW501516 sobre la actividad de unión al ADN de PPARα. La sonda PPRE formó 2complejos principales (i y ii) con las proteínas nucleares hepáticas (fig. 3A). La especificidad de estos 2complejos se comprobó realizando un experimento de competición añadiendo un exceso de sonda PPRE no marcada. La actividad de unión al ADN de PPARα fue mayor en los ratones alimentados con la HFD más GW501516 que en los ratones control y los alimentados con la HFD. Además, el anticuerpo contra PPARα dio lugar a un retardo (supershift) de la migración del complejo ii, indicando así la presencia de PPARα. Por el contrario, no se observó ningún supershift cuando se adicionó un anticuerpo contra PPARβ/δ. En concordancia con el aumento de expresión de Pparα y de su actividad de unión al ADN, el tratamiento con GW501516 incrementó la expresión de los genes diana de PPAR Mcad, Cte y Cpt1a en comparación al control y a los ratones alimentados con la HFD (fig. 3B-D). En concordancia con el aumento de expresión de los genes diana de PPAR implicados en la oxidación de los ácidos grasos, los niveles de β-hidroxibutirato, un producto de la cetogénesis usado como marcador de la oxidación de ácidos grasos hepática, aumentó significativamente en los ratones que recibieron la HFD más el tratamiento con GW501516 (fig. 3E). En conjunto, estos resultados indican que GW501516, al igual que la sobreexpresión de lipina116, es capaz de amplificar la vía PPARα-PGC-1α en el hígado y así aumentar la oxidación hepática de ácidos grasos.
 Autorradiografía de EMSA con sonda PPRE marcada con 32γP y con extractos nucleares. Según la competición con un exceso de sonda no marcada se formaron 2complejos específicos (i y ii). Se realizó análisis de supershift incubando con anticuerpos contra PPARα, PPARβ y Oct1. IC: inmunocomplejo. Efectos del GW501516 sobre los niveles de ARNm de la Mcad (B), la Cte (C) y la Cpt1a (D) y niveles plasmáticos de β-hidroxibutirato (E). Los resultados se expresan como la media±DE (n=5). *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 aumenta la expresión y la actividad de unión al ADN de PPARα y la oxidación de los ácidos grasos en el hígado de ratones alimentados con la HFD. A) Autorradiografía de EMSA con sonda PPRE marcada con 32γP y con extractos nucleares. Según la competición con un exceso de sonda no marcada se formaron 2complejos específicos (i y ii). Se realizó análisis de supershift incubando con anticuerpos contra PPARα, PPARβ y Oct1. IC: inmunocomplejo. Efectos del GW501516 sobre los niveles de ARNm de la Mcad (B), la Cte (C) y la Cpt1a (D) y niveles plasmáticos de β-hidroxibutirato (E). Los resultados se expresan como la media±DE (n=5). *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.
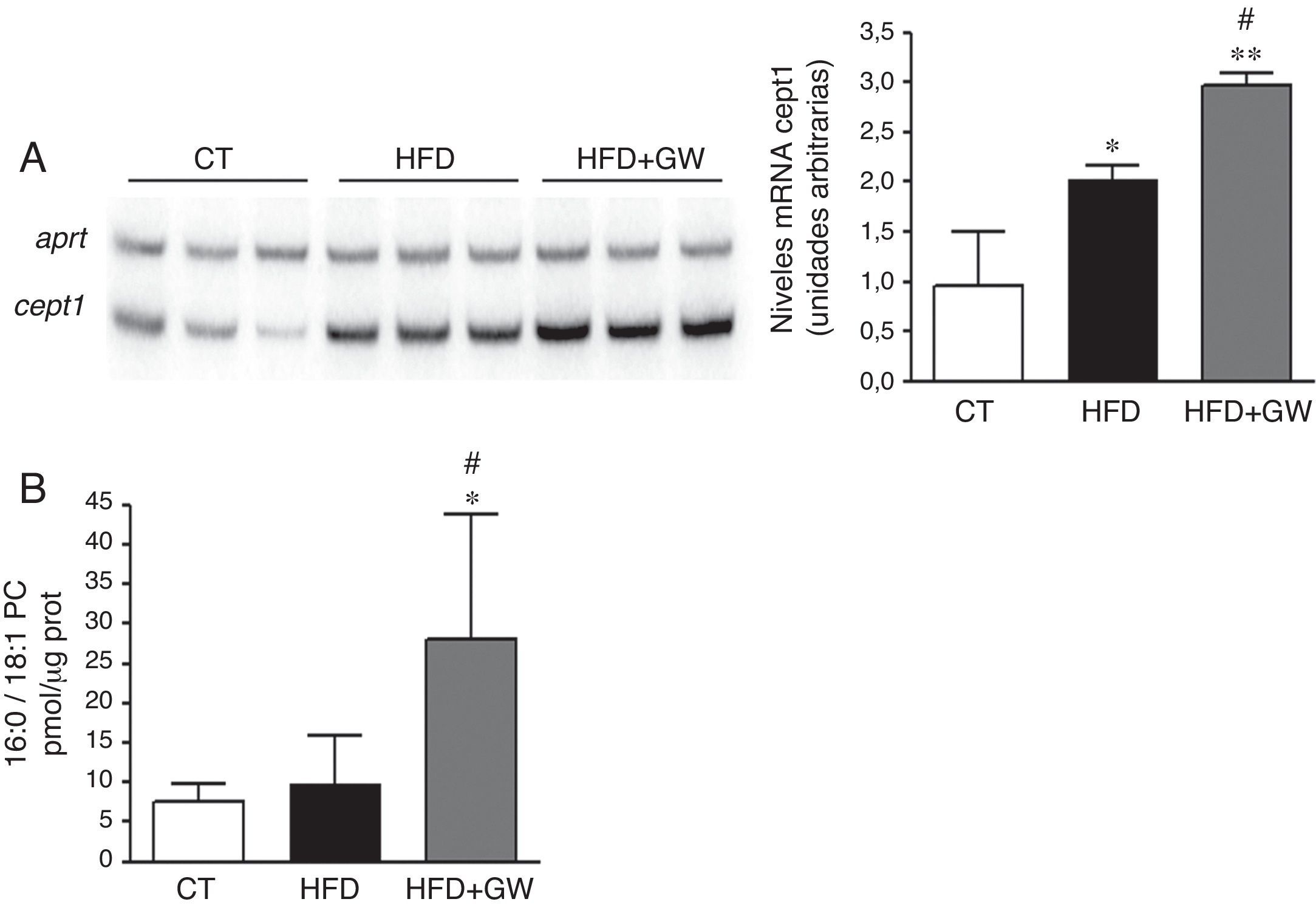
El aumento de la vía PPARα por el tratamiento con GW501516 parecía ser mayor de lo esperado considerando el aumento observado en la lipina1, por lo que decidimos buscar un mecanismo adicional que contribuyera a los efectos causador por GW501516. Recientemente se ha identificado un nuevo ligando endógeno de PPARα, la 16:0/18:1-PC (fosfatidilcolina)26. En estudios de células de hepatoma la sobreexpresión de Cept1, gen que codifica para la enzima responsable de la síntesis de este ligando, produce la activación de PPARα y de sus genes diana como la Cpt1a26. En primer lugar determinamos si el tratamiento con GW501516 había afectado la expresión de Cept1. Tanto la HFD como la HFD más GW501516 aumentaron la expresión de Cept1, pero el aumento atribuido a GW501516 fue mayor que el causado por la HFD sola (fig. 4A). Además, cuando se analizaron los niveles del ligando 16:0/18:1-PC solamente los ratones que recibieron GW501516 mostraron niveles aumentados de este ligando endógeno de PPARα (fig. 4B).
 Niveles de ARNm de la Cept-1. Autorradiografía representativa y cuantificación normalizada con niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5). B) Cuantificación del ligando 16:0/18:1-PC en extractos nucleares de hígado. Resultados expresados como la media±DE (n=5). *p<0,05; **p<0,01 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 aumenta los niveles del ligando endógeno de PPARα 16:0/18:1-PC en ratones alimentados con la HFD. A) Niveles de ARNm de la Cept-1. Autorradiografía representativa y cuantificación normalizada con niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5). B) Cuantificación del ligando 16:0/18:1-PC en extractos nucleares de hígado. Resultados expresados como la media±DE (n=5). *p<0,05; **p<0,01 vs ratones control; #p<0,05 vs ratones alimentados con la HFD.
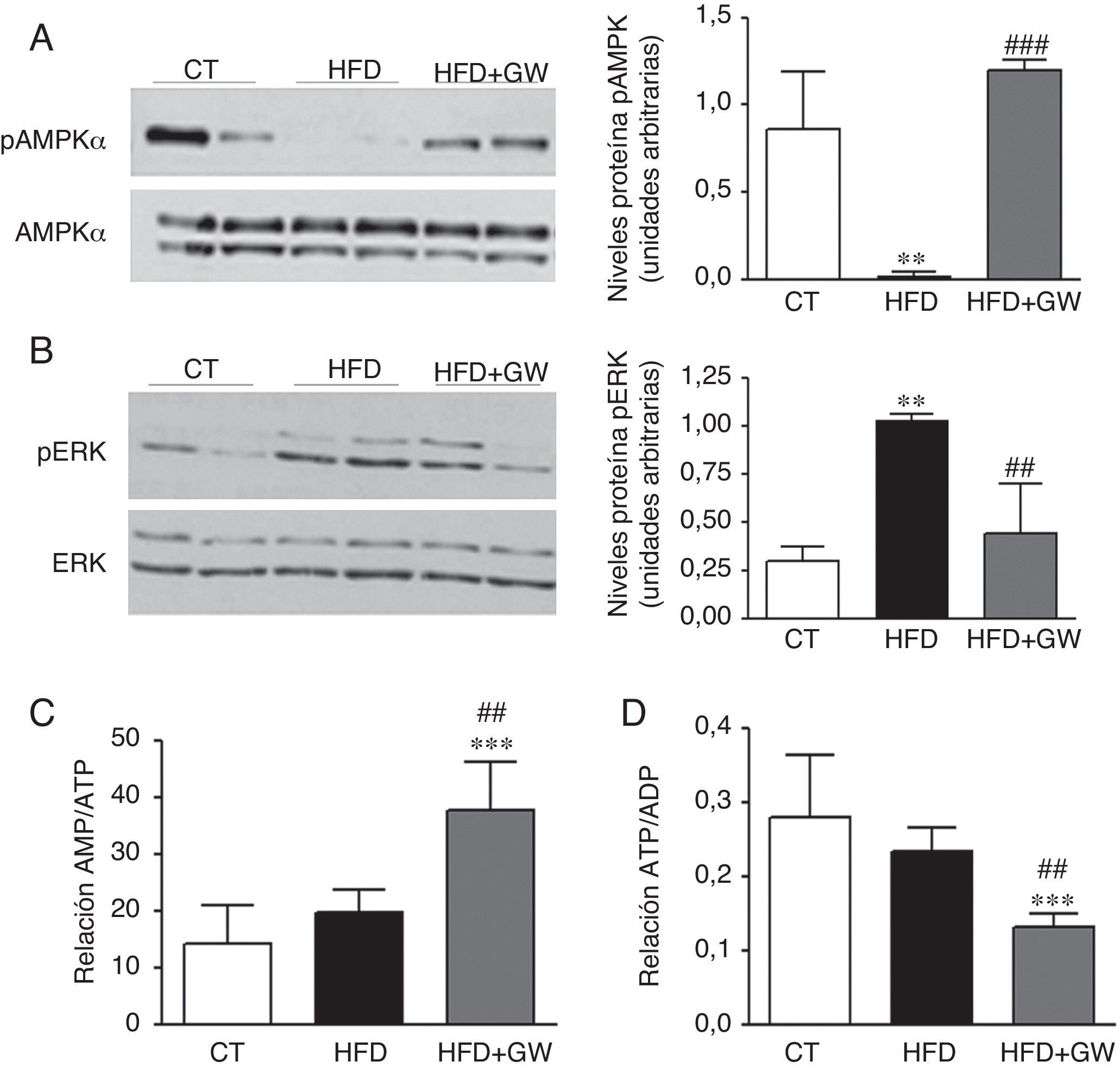
Dado el importante papel regulador de la AMPK en la oxidación de los ácidos grasos en el hígado y el hecho de que esta cinasa puede regular la expresión de lipina125 y Pgc-1α27, analizamos los niveles hepáticos de la AMPK fosforilada y total. De acuerdo con estudios anteriores28, la HFD disminuyó los niveles de la AMPK fosforilada en el hígado (fig. 5A). Además, el tratamiento con GW501516 evitó esta reducción. Se ha descrito que existe una inhibición recíproca entre AMPK y ERK1/229, y que la inhibición de AMPK aumenta la fosforilación de ERK1/2 en el hígado30, por lo que examinamos el estado de esta cinasa. La reducción de la AMPK fosforilada observada en los ratones alimentados con HFD iba acompañada por un aumento en los niveles de ERK1/2 fosforilada, pero este aumento fue atenuado por el tratamiento de GW501516 (fig. 5B). Dado que la AMPK puede ser regulada alostéricamente por AMP31 y está descrito que PPARβ/δ puede interferir en la cadena respiratoria produciendo una reducción de ATP y, por tanto, un aumento de la relación AMP/ATP14, determinamos las concentraciones del nucleótido adenina por cromatografía líquida de alta eficacia para examinar la relación AMP/ATP y ATP/ADP (adenosindifosfato). El tratamiento con GW501516 aumentó la relación AMP/ATP (fig. 5C) en el hígado además de disminuir la relación ATP/ADP (fig. 5D) en comparación a los animales control y los alimentados con la HFD, indicando así que el mecanismo responsable del aumento de la fosforilación de la AMPK inducido por GW501516 se podría deber a un cambio en el estado energético del hepatocito.
 y la ERK1/2 fosforilada (B). Resultados expresados como la media±DE (n=3). Relación AMP/ATP (C) y ATP/ADP (D) en el hígado. Resultados expresados como la media±DE (n=5). *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05; ##p<0,01; ###p<0,001 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 evita la reducción de la AMPK fosforilada e incrementa la relación AMP/ATP. Niveles proteicos de la AMPK fosforilada (A) y la ERK1/2 fosforilada (B). Resultados expresados como la media±DE (n=3). Relación AMP/ATP (C) y ATP/ADP (D) en el hígado. Resultados expresados como la media±DE (n=5). *p<0,05; **p<0,01; ***p<0,001 vs ratones control; #p<0,05; ##p<0,01; ###p<0,001 vs ratones alimentados con la HFD.
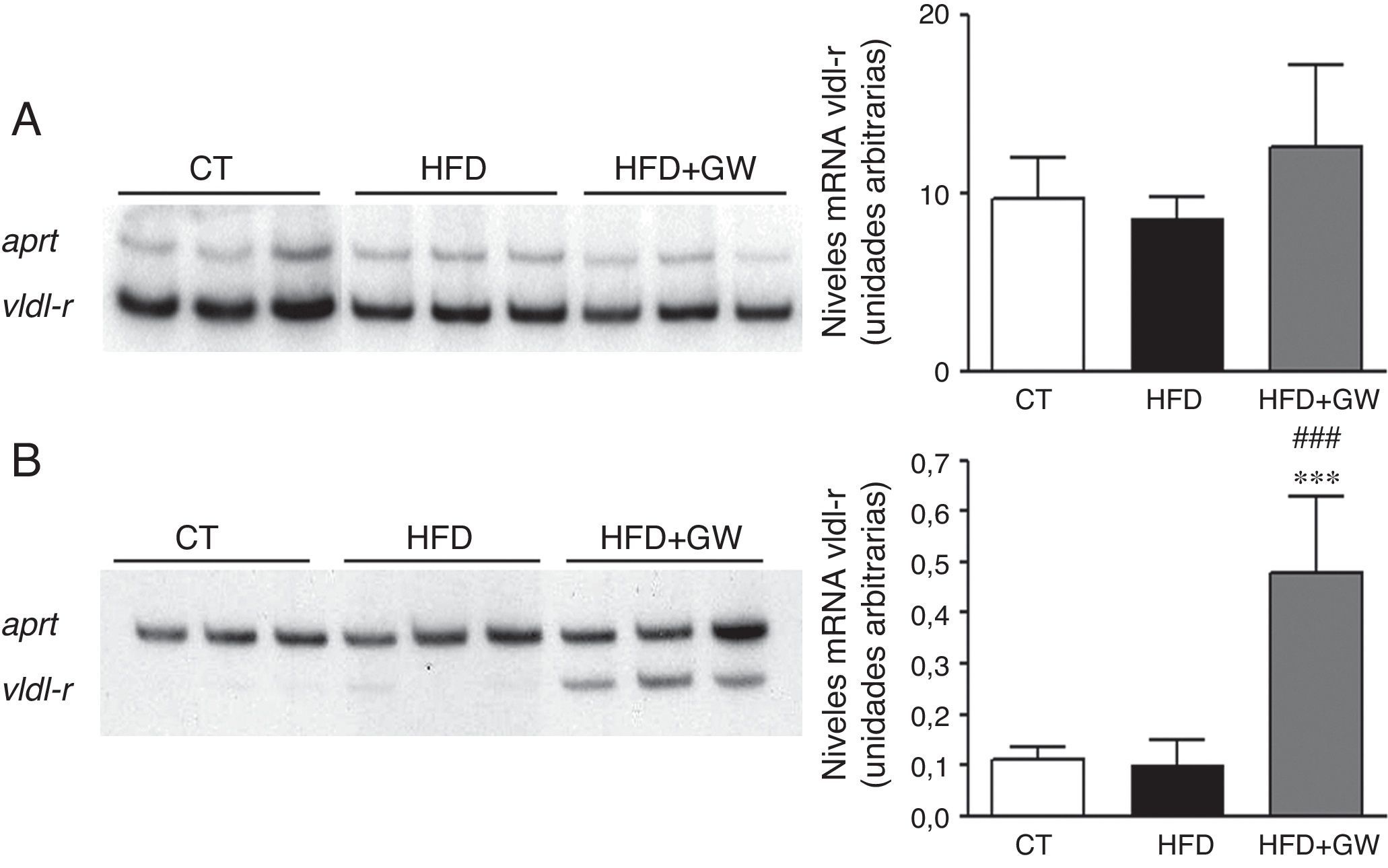
El aumento observado en la oxidación de los ácidos grasos causado por GW501516 muestra una aparente controversia con la acumulación de triglicéridos hepáticos causado por este fármaco. Aunque se ha descrito que esta acumulación podría ser el resultado de un aumento en el flujo de glucosa hacia la vía de las pentosas fosfato y el aumento de la síntesis de ácidos grasos causado por GW5015169, decidimos estudiar si había otros mecanismos adicionales involucrados. Recientemente se ha descrito que el receptor de las VLDL se encuentra regulado por PPARβ/δ32 y que su expresión puede aumentar por acción de la AMPK33. Este receptor se expresa de manera abundante en el corazón, el músculo esquelético y el tejido adiposo, pero solo en trazas en el hígado34. Además, en estudios in vitro se ha demostrado que el receptor de las VLDL une e internaliza lipoproteínas ricas en triglicéridos, incluyendo las VLDL, en el tejido adiposo y en el músculo esquelético, reduciendo así los niveles de triglicéridos plasmáticos35. En concordancia con estos datos, la expresión del receptor de VLDL era muy notable en el músculo esquelético (fig. 6A) y ausente en el hígado (fig. 6B) de los animales control. Sorprendentemente, el tratamiento con GW501516 no afectó la expresión de este receptor en el músculo esquelético, pero sí en el hígado (fig. 6). Estos resultados contribuyen a explicar tanto la reducción de los triglicéridos plasmáticos como la falta de reducción en el contenido de triglicéridos hepáticos tras el tratamiento con GW501516, como resultado de un aumento en la captación de los triglicéridos del plasma.
 y en el hígado (B). Autorradiografía representativa y cuantificación normalizada con niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5).***p<0,001 vs ratones control; ###p<0,001 vs ratones alimentados con la HFD.")
El tratamiento con GW501516 aumenta los niveles de ARNm del receptor de las VDL en el hígado. Niveles de ARNm del receptor de las VLDL en el músculo esquelético (A) y en el hígado (B). Autorradiografía representativa y cuantificación normalizada con niveles de ARNm del gen control Aprt. Los resultados se expresan como la media±DE (n=5).***p<0,001 vs ratones control; ###p<0,001 vs ratones alimentados con la HFD.
El consumo excesivo de grasas puede producir un desequilibrio en el metabolismo lipídico hepático provocando un incremento de la producción y liberación de lipoproteínas, alteración asociada al desarrollo de trastornos metabólicos como la obesidad, la resistencia a la insulina y el síndrome metabólico. La utilización del agonista de PPARβ/δ GW501516 ha demostrado en diversos estudios animales10, e incluso en humanos36, un efecto hipotrigliceridemiante de este fármaco, lo que sugería que podría ser útil para el tratamiento de estos trastornos. En este estudio se evaluó el efecto del activador de PPARβ/δ GW501516 sobre su capacidad de regular la oxidación de los ácidos grasos en el hígado de ratones alimentados con una dieta rica en grasas (HFD). Nuestros resultados demostraron que el tratamiento con GW501516 previene el desarrollo de hipertrigliceridemia causada por la HFD. Además, estos datos muestran que el tratamiento con este activador de PPARβ/δ previene la reducción hepática de los niveles de la AMPK fosforilada inducida por la HFD y aumenta la oxidación de los ácidos grasos en el hígado por amplificación de la actividad de la vía lipina1-PPARα-PGC-1α.
A diferencia de la mayoría de los estudios donde la exposición a la HFD es a más largo plazo (14-18semanas), en nuestro estudio los ratones fueron alimentados con una HFD durante un período corto (3semanas) para observar mejor los cambios iniciales que producen el desequilibrio metabólico causado por esta dieta. La administración de la HFD provocó en los ratones hipertrigliceridemia e intolerancia a la glucosa, pero no se observaron cambios significativos en los niveles plasmáticos de ácidos grasos libres, leptina o adiponectina, hecho que indicaba que en nuestras condiciones la HFD no indujo señales derivadas del tejido adiposo que interfirieran sobre el metabolismo hepático. En cambio, los resultados mostraron que el tratamiento con GW501516 era capaz de evitar el desarrollo de la hipertrigliceridemia causada por la HFD. Además, se observó una mejora en la sensibilidad a la insulina en los ratones tratados con el agonista de PPARβ/δ.
Los ratones alimentados con la HFD mostraron una reducción de los niveles hepáticos de la AMPK fosforilada que se evitó por la administración de GW501516. El mantenimiento de la AMPK fosforilada fue acompañado por la recuperación de los niveles de expresión génica hepáticos de lipina1 y Pgc-1α y por el aumento de los niveles de ARNm del receptor de VLDL. Aunque no se puede descartar un efecto directo en la activación transcripcional de estos genes por PPARβ/δ, ya que se ha descrito que lipina1, el receptor de las VLDL y Pgc-1α son genes diana de PPARβ/δ, la mayoría de los efectos de GW501516 serían atribuibles al aumento de la fosforilación de la AMPK14. De hecho, se ha descrito que esta cinasa puede regular la expresión de lipina125, el receptor de las VLDL33 y Pgc-1α27. Por otro lado, se ha descrito una inhibición recíproca entre ERK1/2 y AMPK29, por lo que el aumento en los niveles de la AMPK fosforilada podría ser el resultado de la inhibición de la fosforilación de ERK1/2 causada por GW501516, resultado que coincide con estudios previos donde GW501516 prevenía la fosforilación de ERK1/2 inducida por LPS en adipocitos37. Es importante resaltar que un estudio anterior muestra que la presencia de obesidad permite el aumento de actividad en la ERK1/2 en el hígado y que la restricción calórica inhibe este aumento y mejora la sensibilidad a la insulina38. En nuestro estudio la mejoría en la tolerancia a la glucosa causada por GW501516 también iba acompañada de una reducción en los niveles de fosforilación de la ERK1/2.
Asimismo, en este estudio mostramos que GW501516 aumenta la relación AMP/ATP en el hígado, indicando que, de acuerdo a un estudio anterior realizado en células de músculo esquelético14, el aumento de la fosforilación de la AMPK se debería a un cambio en el estado energético de la célula. Estudios previos sugieren que la reducción de los niveles de ATP causado por GW501516 podría deberse a una inhibición específica de uno o más complejos de la cadena respiratoria, un efecto sobre la síntesis de ATP o el desacoplamiento mitocondrial14, cambios que reducirían los niveles de ATP generados por la mitocondria y permitirían la activación de la AMPK.
Como se ha comentado anteriormente, la activación de la AMPK puede regular la expresión de lipina1 y Pgc-1α. Además, la lipina1 puede actuar como co-activador transcripcional interaccionando directamente con los receptores nucleares PPARα y PGC-1α16, activando así genes implicados en la fosforilación oxidativa mitocondrial y de los ácidos grasos. En nuestro estudio el tratamiento con GW501516, en consonancia con la recuperación de los niveles de la AMPK, evitó la reducción de PGC-1α y aumentó los niveles de lipina1 nuclear, permitiendo la amplificación de la vía PGC-1α-PPARα, hecho que se demuestra tanto por el aumento de la expresión génica como de la actividad de unión al ADN de PPARα. Además, el tratamiento con GW501516 aumentó la expresión de una serie de genes diana de PPARα implicados en la oxidación de los ácidos grasos, como son la Mcad, la Cte y la Cpt1a. Asimismo, el tratamiento farmacológico aumentó los niveles plasmáticos de β-hidroxibutirato, uno de los productos finales de la β-oxidación hepática. Por otro lado, la activación de PPARα también podría ser consecuencia de un aumento de los niveles de sus ligandos endógenos como es el 16:0/18:1-PC, el cual presentaba niveles nucleares aumentados en los animales tratados con GW501516. Finalmente, todos estos resultados muestran que el agonista de PPARβ/δ GW501516 previene la reducción de la AMPK, de PGC-1α y de la lipina1, amplifica la vía PPARα y aumenta la oxidación de los ácidos grasos, cambios que pueden contribuir a reducir la secreción hepática de triglicéridos y, en consecuencia, los niveles de triglicéridos plasmáticos.
Sorprendentemente, en contradicción aparente con al aumento de la oxidación de los ácidos grasos, los animales tratados con GW501516 presentaban una acumulación hepática de triglicéridos similar a la observada en los animales que solo recibieron la HFD. Esta acumulación de lípidos se podría explicar por diversas razones. En primer lugar se ha demostrado que la sobreexpresión hepática de lipina1 reduce la secreción de triglicéridos secuestrándolos en el hígado16. Por otro lado, como ya se ha comentado anteriormente, el tratamiento con GW501516 aumentó los niveles del receptor de las VLDL. Este aumento podría contribuir al efecto hipotrigliceridemiante del GW501516, ya que podría aumentar la captación de VLDL del plasma, retirándolas de la circulación sanguínea para su almacenaje u oxidación en el hígado. Finalmente, otro hecho que explicaría la acumulación de triglicéridos hepáticos podría ser la capacidad de GW501516 para inducir la síntesis de ácidos grasos a partir de glucosa en el hígado, tal y como ha sido demostrado por Lee et al.27 A pesar de ello, según este mismo estudio, este efecto podría deberse a la duración del tratamiento, ya que no se observó hígado graso en un tratamiento de 6meses con GW501516. Así pues, se podría pensar que a largo plazo la oxidación de los ácidos grasos hepática activada por GW501516 sería capaz de reducir tanto los triglicéridos circulantes como los acumulados en el hígado. Además, recientemente se ha demostrado que la activación de PPARβ/δ en el hígado produce acumulación de lípidos mayoritariamente en forma de ácidos grasos monoinsaturados en detrimento de los ácidos grasos saturados mucho más lipotóxicos39.
Responsabilidades éticasDerecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Protección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
FinanciaciónEste estudio se financió parcialmente por el Ministerio de Economía y Competitividad (SAF2009-06939 y BFU2010-18826) y por el Centro de Investigación Biomédica en Red Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), una iniciativa del Instituto de Salud Carlos III.
AutoríaEmma Barroso y Manuel Vázquez-Carrera se encargaron del diseño de los experimentos, de la recogida de datos y de la interpretación de estos, así como de la redacción, la revisión y la aprobación del contenido del artículo. Alma Astudillo y Jesús Balsinde recogieron datos y contribuyeron a su interpretación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Una comunicación referente a esta línea de trabajo, titulada «La activación de PPARβ/δ previene la hipertrigliceridemia causada por una dieta rica en grasas. Implicación de la AMPK y de la vía PGC-1α-lipina 1-PPARα», fue presentada en el XXIV Congreso Nacional de la SEA (Sevilla 2011) y fue galardonada con una mención especial.