







Las enfermedades raras, pese a su baja frecuencia individual, afectan globalmente al 7% de la población, por lo que el profesional de Atención Primaria (AP) tendrá varios de estos pacientes bajo seguimiento. El 80% de estas enfermedades tienen base genética, lo que hace fundamental un asesoramiento genético adecuado. El seguimiento de pacientes con síndrome de Wolfram (SW) puede servir para diseñar un protocolo susceptible de ser utilizado en el diagnóstico y manejo de otras entidades y ser utilizado por profesionales sanitarios para dar soporte a los pacientes, contando con la participación de profesionales sanitarios e investigadores especializados en el SW, los propios pacientes y su entorno. Se desarrollan los pasos fundamentales de todo procedimiento clínico genético, en el que la AP es clave para dar soporte a estas familias y transmitir de forma comprensible la información sobre los aspectos genéticos.
Rare diseases, despite their individual low frequency, affect 7% of the population all combined. Consequently, every primary care practitioner (PCP) will have several of these patients under his care. 80% of rare diseases are genetically determined, which makes genetic counseling fundamental in these cases. The follow-up of patients with Wolfram syndrome (WS) can be used to design a protocol to support these patients, with the participation of researchers and healthcare professionals specialized in WS, the patients themselves and their familial environment. This protocol can be suitable for the diagnosis and management of other diseases as well. The main steps of every genetic clinical procedure are developed in this article, emphasizing the role of PCP in supporting patients and their families and in transmitting genetic information in a comprehensible manner.
Las enfermedades raras (ER) o poco frecuentes −este último término es cada vez más usado, debido a las posibles connotaciones negativas de la palabra «raro»− constituyen un grupo heterogéneo y numeroso de patologías que, a pesar de su baja prevalencia individual, suponen un importante problema sanitario. No disponemos de una definición válida a nivel global, sino que en cada región se utilizan criterios distintos para su determinación. En Europa, por ejemplo, se define como ER a aquella que no afecta a más de 5 de cada 10.000 personas (1/2.000)1, mientras que en Japón toman como punto de corte una prevalencia de 1 de cada 2.500 personas2.
Esta definición basada en la prevalencia es insuficiente, ya que no tiene en cuenta las repercusiones a nivel social y sanitario. Es por ello que la Unión Europea tiene en cuenta también otros factores determinantes como la gravedad de la enfermedad y su morbimortalidad, la afectación de la calidad de vida o la inexistencia de tratamientos adecuados.
Actualmente se han identificado entre 6.000 y 8.000 ER distintas. A pesar de conformar un grupo muy heterogéneo que engloba entidades muy diversas, comparten una baja prevalencia, elevada morbimortalidad y un carácter progresivo. A menudo debutan en la infancia y hasta un 80% tiene una base genética. Todo ello repercute en unas carencias/necesidades socio-psico-sanitarias similares3.
Se ha estimado que la prevalencia global de las ER en la Unión Europea se sitúa en torno al 7%, lo que supone en números absolutos entre 24 y 36 millones de personas, tres de los cuales corresponderían a España4. Por las características descritas anteriormente, estas entidades no solo afectan al propio paciente, sino a toda su familia y entorno, máxime teniendo en cuenta que la mayoría debutan en la infancia, haciendo más evidente el gran desajuste familiar que producen. Es, por tanto, oportuno considerarlas como un problema de salud pública5,6.
Se tiende a pensar que las ER con base genética se manifiestan con un fenotipo característico que facilite el proceso diagnóstico. Sin embargo, no siempre es posible establecer una relación sencilla entre el fenotipo clínico y las alteraciones genéticas causales. Aun así, la carga genética en las ER puede ser el punto de partida para recabar información acerca del origen o el desarrollo de la patología. Asimismo, numerosos tratamientos potenciales y en desarrollo se basan en la información genética asociada a una enfermedad5.
La atención a estos pacientes, que implica establecer una sospecha clínica, realizar el diagnóstico y proporcionar un seguimiento y un consejo genético adecuados, suele involucrar a diversos especialistas médicos. La participación del médico de Atención Primaria (AP) en el diagnóstico y consejo genéticos tradicionalmente ha sido escasa. Sin embargo, la difusión de los estudios moleculares y el aumento de su disponibilidad hacen evidente la necesidad de que el médico de AP tome parte activa en este proceso, actuando como puente entre el paciente y el especialista en genética7,8.
Desafortunadamente, no hay consenso aún sobre las funciones exactas que este profesional debería desempeñar dentro de la estructura de diagnóstico y consejo genético, siendo fundamental la formación en genética, que en muchos casos se percibe como deficitaria9. No obstante, la tendencia actual es al aumento de la participación del profesional de AP, especialmente en casos de menor complejidad10.
Por este motivo, y con la intención de fomentar el papel de la AP en este campo, se describirá a continuación el protocolo a seguir durante el proceso asistencial a pacientes con ER desde el punto de vista genético, destacando las funciones del profesional de AP en este proceso −desde la sospecha diagnóstica hasta el consejo genético tras el diagnóstico confirmatorio−. Para ello, en este trabajo vamos a usar como ejemplo el SW, dado que cumple con todas las características que definen por qué es tan importante un adecuado asesoramiento genético en estas entidades: baja prevalencia, morbimortalidad importante con repercusiones psicológicas asociadas y etiología genética.
Para la elaboración de este protocolo se ha requerido la participación de diferentes profesionales sanitarios e investigadores especializados en diversos aspectos de la misma y se ha contado con la colaboración activa de las personas que padecen la enfermedad, sus familiares y su entorno como pieza fundamental en todas las fases del proceso de elaboración. En el SW, y en general en todas las ER, la implicación de pacientes y profesionales sanitarios resulta esencial para conseguir que el protocolo de actuación sea plenamente satisfactorio y responda a las necesidades de todos adecuadamente.
Síndrome de WolframEl SW es una compleja enfermedad neurodegenerativa que se engloba dentro de las ER, con una prevalencia global de una de cada 770.000 personas11. Se asocia con una gran variedad de síntomas, debutando generalmente en la primera década de vida, y dicha heterogeneidad clínica depende de la penetrancia del gen afectado12. Sin embargo, sus cuatro manifestaciones clínicas principales son la diabetes mellitus (DM), atrofia óptica (OA), sordera neurosensorial (deafness) y diabetes insípida (DI), que dan lugar al acrónimo DIDMOAD, por el que también se conoce esta enfermedad13,14. Algunos autores apuntan a la necesidad de incorporar también las alteraciones urinarias como componente principal del SW y denominarlo DIDMOADUA12,13.
En la tabla 1 podemos ver los criterios diagnósticos del SW, elaborados por la guía clínica del manejo del SW12.
Criterios diagnósticos de síndrome de Wolfram. Porcentajes en paréntesis referidos a la prevalencia del registro de EURO-WABB14 (121 participantes con diagnóstico genético confirmado)
| Criterios mayores | Criterios menores | Mínimo requerido | Otras variables sugestivas de evidencia |
|---|---|---|---|
| - Diabetes mellitus < 16 años (87%)- Atrofia óptica < 16 años (80%) | - Diabetes insípida (42%)- DM >16a (4%)- Atrofia óptica > 16a (7%)- Sordera neurosensorial (48%)- Signos neurológicos (ataxia, epilepsia, deterioro cognitivo (29%)- Anormalidades tracto renal (estructural o funcional) (33%)- Una mutación con pérdida de función en WFS1/CISD2 y/o antecedentes familiares de SW | - Dos criterios mayoreso- Uno mayor y dos menoreso- Dos mutaciones patológicas identificadas en WFS1 o CISD2 | - Hipogonadismo (hombres) (6%)- Ausencia de Autoanticuerpos DM tipo 1- Cataratas bilaterales (1%)- Alteraciones psiquiátricas (26%)- Alteraciones gastrointestinales (5%) |
DM: diabetes mellitus; SW: síndrome de Wolfram.
El SW está asociado a mutaciones en dos genes distintos, lo que da lugar dos tipos bien diferenciados: el tipo 1 (SW1) y el tipo 2 (SW2). Los genes involucrados son WSF1 (4p16.1) en el SW1 y CISD2 (4q24) en el SW212.
El gen WFS1 codifica la proteína wolframina, que se expresa de manera ubicua en el retículo endoplásmico de todas las células, pero tiene una alta expresión en las células beta de los islotes pancreáticos y en las neuronas cerebrales. El gen CISD2 codifica la proteína 2 CDGSH, que se encuentra también en el retículo endoplásmico, pero no interacciona directamente con la wolframina. Las mutaciones en WFS1 suelen ser deletéreas para la expresión proteica, conduciendo a una falta de función de la misma y a un alto nivel de estrés del retículo endoplásmico por pérdida de calcio hacia el citoplasma, lo que activa una respuesta de estrés en el citoplasma que culmina en apoptosis celular12,15.
Existen diferencias fenotípicas entre ambos tipos de SW. Mientras que en el SW1 padecen diabetes insípida, los pacientes afectos de SW2 no la padecen, pero sufren de ulceraciones y sangrados en la parte alta del tracto gastrointestinal16,17.
El patrón de herencia es autosómico recesivo, si bien en algunos casos aislados se ha relacionado con herencia mitocondrial12. Asimismo, mutaciones de WSF1, esta vez en heterocigosis, han sido asociadas con formas autosómicas dominantes para algunos síntomas del SW, como la pérdida auditiva neurosensorial no sindrómica de baja frecuencia DFNA6/14/38 (LFSNHL)18 atrofia óptica, disminución auditiva, diabetes tipo 2 y problemas psiquiátricos13. Además, hay descritas otras mutaciones asociadas al SW, que manifiestan una sintomatología y grado de alteración diferente en cada caso12,15,17.
Al hablar de herencia autosómica recesiva, se entiende que es necesario poseer los dos alelos mutados del gen involucrado para poder padecer la enfermedad. Esta condición puede ser resultado de heredar de cada progenitor un alelo mutado (lo más habitual), o bien de heredar únicamente un solo alelo mutado de alguno de los dos padres y producirse una segunda mutación de novo en el propio paciente. Los individuos que solo poseen un alelo mutado (portadores) no presentan síntomas de la enfermedad, pero pueden transmitirla a su descendencia12.
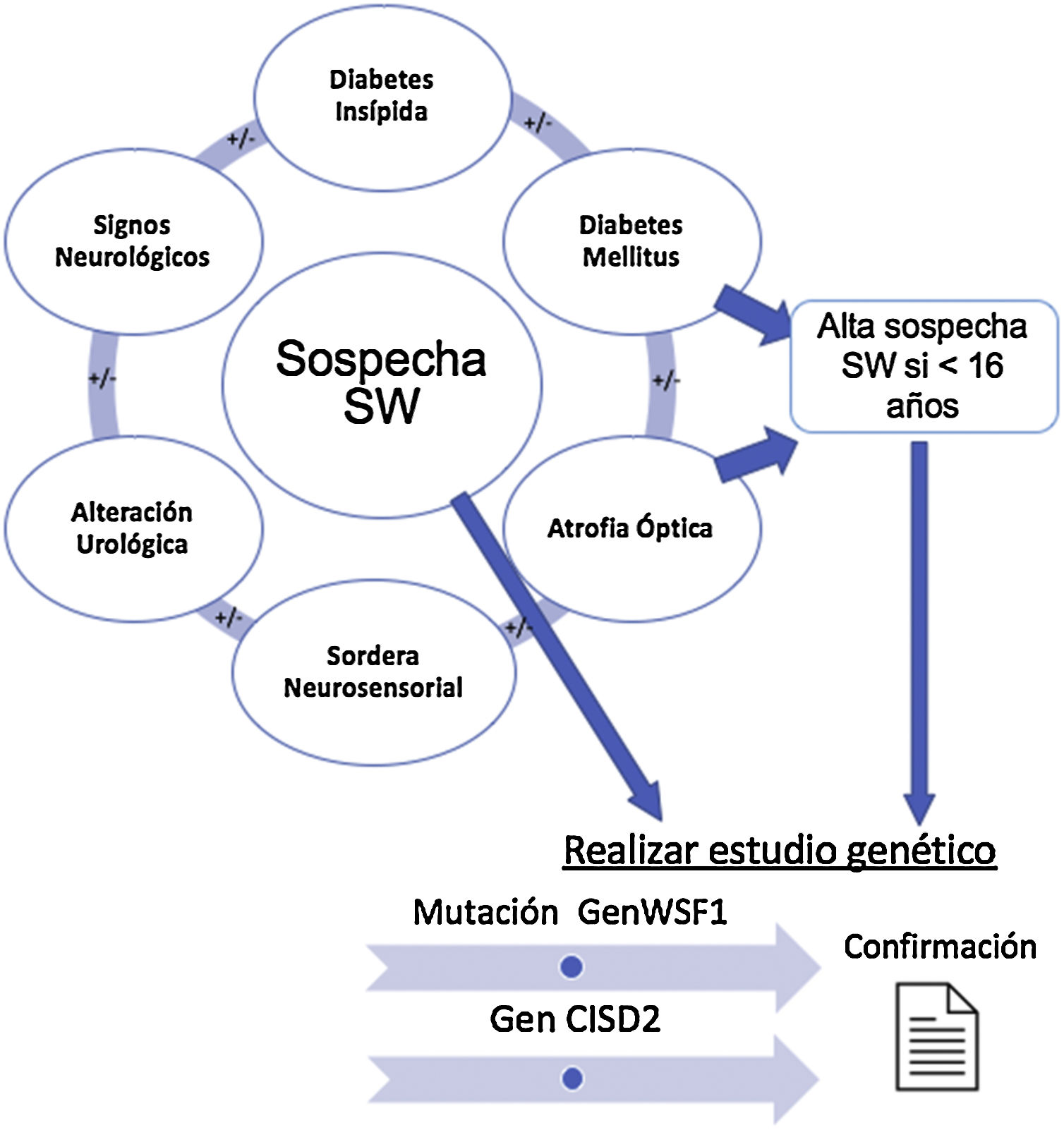
La figura 1 resume los hallazgos clínicos que alertan sobre la posible existencia de un SW, cuya presencia nos llevará a solicitar el estudio genético confirmatorio.

Establecimiento de la sospecha clínica de síndrome de Wolfram y solicitud de estudio genético. Diabetes mellitus y diabetes insípida, atrofia óptica y sordera son los componentes básicos. En presencia de signos clínicos compatibles, se solicitará el estudio de los genes WSF1 y CISD2 para confirmar el diagnóstico.
El conocimiento de la base genética de una enfermedad, en este caso del SW, permite, además de realizar el diagnóstico y caracterizar la enfermedad en el propio paciente, realizar un consejo genético adecuado a los familiares, informando de la probabilidad de transmitir a su descendencia los alelos alterados y de que padezcan la enfermedad15.
La consulta de genética debe estar dirigida a obtener información relevante sin pasar por alto los derechos del usuario. El médico de AP debe formar parte de la estructura de diagnóstico genético a lo largo de todo el proceso y su papel es clave para una buena intervención de salud en materia de genética19, como se irá detallando más adelante. La información, aunque haya sido ofrecida previamente por un especialista en genética clínica, se verá sin lugar a duda reforzada y mejor comprendida si el médico de AP asume una parte activa en el proceso, pues se trata del médico más accesible para el paciente y sus familiares. A continuación, se detallan cuatro pasos fundamentales que debe cumplir todo procedimiento clínico genético.
Identificación de individuos o familias de riesgoEl primer paso consiste en seleccionar a los pacientes que, por hallazgos clínicos, radiológicos o analíticos, tengan más probabilidades de padecer una ER. El médico de AP, como puerta de entrada en el sistema sanitario de la población general, es el que habitualmente marca la sospecha diagnóstica de ER. Se deberá realizar una anamnesis por aparatos y una exploración física detallada, incluyendo datos de otras patologías que puedan estar relacionadas y nos ayuden a encuadrar al individuo en un síndrome concreto.
Además de la historia clínica del individuo de riesgo, es clave la confección del árbol genealógico familiar, que puede orientar hacia la posibilidad de que el paciente padezca un síndrome hereditario. Es importante tranquilizar al paciente y a su familia para que entiendan, en este primer momento, que se trata de una sospecha clínica y que necesita una confirmación. La mejor manera de proporcionar serenidad en este proceso es que el profesional se brinde a acompañar a la familia y en este punto es primordial nuevamente el papel de la AP.
Será necesario posteriormente solicitar consulta a un especialista de genética para obtener el diagnóstico confirmatorio20. En este punto nos gustaría reivindicar la importancia de que, ante la alta sospecha de un síndrome de base genética, el médico de AP pueda solicitar cita al especialista oportuno, incluyendo al genetista21.
Consejo genético pretest y firma del consentimiento informadoEn la etapa del consejo genético pretest se proporciona información sobre los riesgos, beneficios y limitaciones del estudio genético, para el cual será necesaria una extracción de sangre y la firma previa de un consentimiento informado para el análisis genético de la muestra. Esta fase se realiza en la consulta especializada.
Con carácter general, el beneficio principal del estudio es el diagnóstico precoz y los riesgos asociados son los trastornos psicológicos derivados de un posible diagnóstico de una enfermedad grave en la que no suele haber tratamientos eficaces, como el SW. Las limitaciones incluirían la posibilidad de un resultado no concluyente y, por tanto, la falta de un diagnóstico definitivo. Hay un alto porcentaje de enfermedades ER que se enfrentan a esta situación, lo cual genera un estrés importante que el profesional de AP debe saber manejar y orientar22.
Información de los resultados del estudio genéticoTras obtener los resultados del estudio, lo ideal es que especialista de genética se reúna con familia y paciente para trasladar de forma adecuada esta información. Se comentarán los aspectos clínicos, moleculares y de manejo médico para que comprendan hasta qué punto la ciencia puede ayudar a convivir con esa enfermedad23. Lo habitual es que en esa consulta se traten temas muy preliminares y se reserve la planificación del programa definitivo de manejo y tratamiento para la siguiente visita.
A los familiares se les explicará la posibilidad de padecerla y se les orientará sobre la necesidad de realizar o no estudios genéticos en los distintos miembros de la familia y también se informara sobre la posibilidad de que otros miembros no presentes se encuentren en riesgo, indicando el interés de contactar con ellos para darles un asesoramiento adecuado.
Tras esta consulta, la familia suele presentar múltiples inquietudes, dudas y miedos, por lo que la orientación de su médico de AP propicia una mejor asimilación de esta información.
Seguimiento y consejo genético postestEl proceso no debe cerrarse tras dar en la anterior visita el diagnóstico. Es preciso realizar una nueva visita, pasado un tiempo de la anterior, con el propósito de que puedan procesar toda la información. En esta visita es importante conocer en la familia y afectado, el impacto emocional de este diagnóstico confirmatorio y las dudas que se han generado para poder ofrecerles el apoyo necesario. En esta visita, en la que la familia ya ha analizado la información, se pueden exponer las medidas a tomar según las opciones informadas por el médico de AP y el especialista de genética.
El profesional de AP debe tener presente la necesidad de evaluación psicológica de la familia y afectados, que habrá de llevarse a cabo durante todo el proceso. Es interesante que se establezca un seguimiento del paciente y su familia a nivel emocional y no cerrar el proceso, dado que se trata de una enfermedad con la que convivirán toda la vida. Además, durante las distintas etapas vitales, sin lugar a dudas, se les plantearán dudas sobre la posibilidad de transmitir esta patología a su descendencia y sobre las oportunidades que los nuevos avances genéticos en el campo de la fertilidad o de su enfermedad les ofrecerán.
Circunstancias y condiciones particularesEl procedimiento descrito anteriormente tiene en cuenta las pautas generales de la consulta de consejo genético24. No obstante, tanto el SW como otras patologías requieren considerar aspectos que pueden ser notables a nivel individual. A continuación, se incluyen una serie de reseñas concretas relacionadas con la cuarta fase del consejo genético postest que se deben tener en cuenta, según la situación particular de cada individuo o familia (fig. 2), y que se explican más adelante:
Familiar implicado
En todas las fases del consejo genético será necesario tener en cuenta a la familia, sobre todo cuando los pacientes sean menores de edad.
El familiar o cuidador es un elemento primordial en la humanización de la atención que se lleva a cabo en el hospital. Cuando un miembro de la familia enferma, se afecta a la familia en su estructura y funcionamiento; cambian las prioridades y valores de la misma y se prioriza la recuperación o mejora en la calidad de vida de la persona que ha enfermado6.
Será por ello imprescindible que durante todo el proceso se trabaje reconociendo a la familia como la unidad de cuidado para que así reciba un apoyo sostenido, personalizado y oportuno, evitando que «se sientan abandonados» y se agoten sus recursos y capacidades. Aquí juega un papel muy importante el médico de AP.
Edad del individuo diagnosticadoLo ideal es que el SW se diagnostique en edad pediátrica, por lo que la información ha de ofrecerse a los progenitores/tutores del afectado. Y con su consentimiento sería además deseable poder explicarle esta información en lenguaje sencillo al afectado en torno a los 14 años, que es cuando realiza la transición de ser atendido por los pediatras a ser atendido por los profesionales de adultos.
Privacidad del afectadoEn el caso de que diagnostiquemos de SW a un paciente en edad adulta, solo él puede decidir cuándo y cómo hablar sobre su enfermedad con sus familiares y amigos. No obstante, si decide hacerlo, se brindará a la familia apoyo psicológico y consejo para sobrellevar su nueva situación vital y, llegado el momento, el cuidado adecuado del enfermo. De igual forma, se debe respetar su derecho a recibir y aceptar los tratamientos pertinentes recomendados.
Si el diagnóstico resulta positivo en un paciente menor de edad, será responsabilidad de los padres (que no estuvieran privados de la patria potestad) o del representante legal del menor, asumir la función de presunta voluntad del mismo tanto para recibir los tratamientos pertinentes, como de acompañamiento en todo el proceso. En este caso también se concederá a la familia apoyo psicológico y consejo para sobrellevar esta nueva situación y para el cuidado adecuado del enfermo.
Asesoramiento reproductivoLos afectados, cuando llegan a edad reproductiva, pueden manifestar su intención de tener descendencia. La consulta de consejo genético debe aportar información suficiente al individuo sobre estos aspectos. Es fundamental trabajar conjuntamente para plantear y valorar sus opciones reproductivas, que dependerán de cada situación concreta.
Las alternativas reproductivas de los afectados como de los progenitores de un afectado deben estudiarse individualmente con el mayor rigor posible, aportando de forma detallada toda la información disponible.
ConclusionesLas ER, por sus características de elevada morbimortalidad, escasez de tratamientos y evolución, por lo general progresiva, conllevan importantes repercusiones psicológicas en los pacientes afectados y su entorno6. Estas características hacen imprescindible, en las ER con base genética, el acceso a un consejo genético adecuado para los individuos afectos. Si bien se ha tomado el SW como ejemplo, el contenido de este texto y las ideas presentadas son también aplicables a las demás ER de etiología genética, pues como se ha dicho anteriormente, presentan características communes, a pesar de su heterogeneidad clínica y genética.
En este artículo se han descrito con detalle las características de un buen asesoramiento genético para pacientes y familias con SW y los pasos que se deben llevar a cabo al realizarlo. El objetivo de dicho asesoramiento será el de proporcionar al paciente y su entorno la mejor atención posible y ayudarlos a convivir con la enfermedad. Un procedimiento de consejo genético, correctamente validado por especialistas y pacientes con SW, permite mejorar la detección, diagnóstico y seguimiento de los afectados. Además, proporciona pautas generales extensibles para muchas otras enfermedades con base genética.
Como se ha expuesto, la participación activa del médico de AP, debido a las características propias y únicas de esta especialidad, y su colaboración con el especialista en genética clínica resultan indispensables a lo largo de todo este proceso.
Por todos estos motivos, los autores pretenden reivindicar la figura del médico de AP en el diagnóstico, seguimiento y consejo genético de pacientes con enfermedades raras que indudablemente resultará en la mejora de la calidad asistencial.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
A todas las familias de la Asociación Española para la Investigación y Ayuda al Síndrome de Wolfram que nos han mostrado su realidad y han hecho posible este trabajo.
A todos los profesionales del equipo multidisciplinar español del Síndrome de Wolfram que día a día aporta esperanzas a las familias.
A Nuria Guirado Romero por la revisión de este trabajo
