




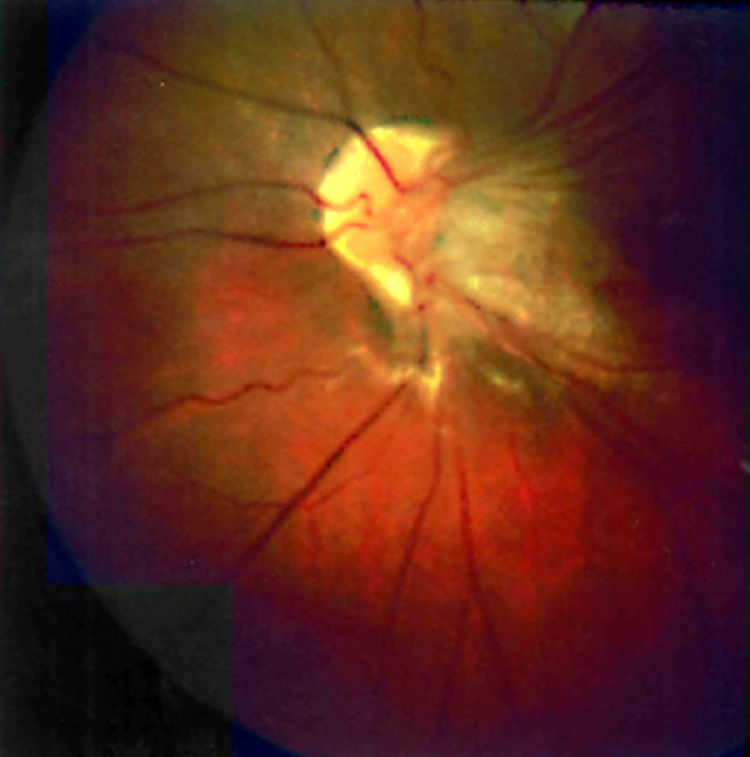
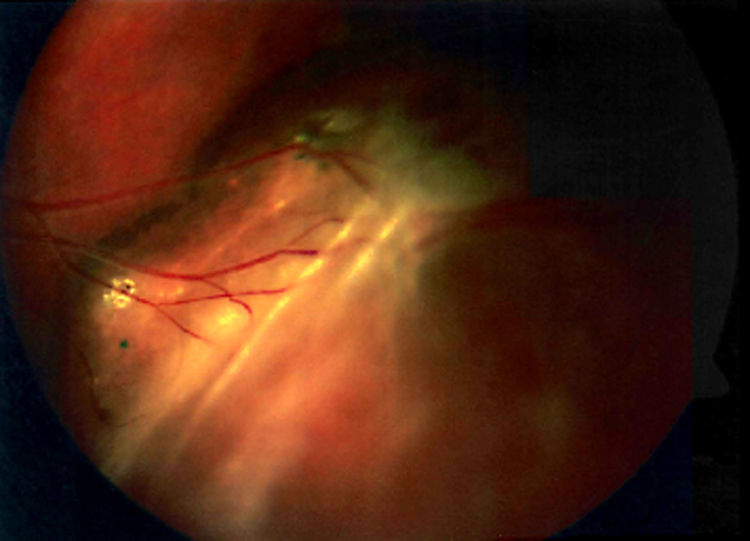
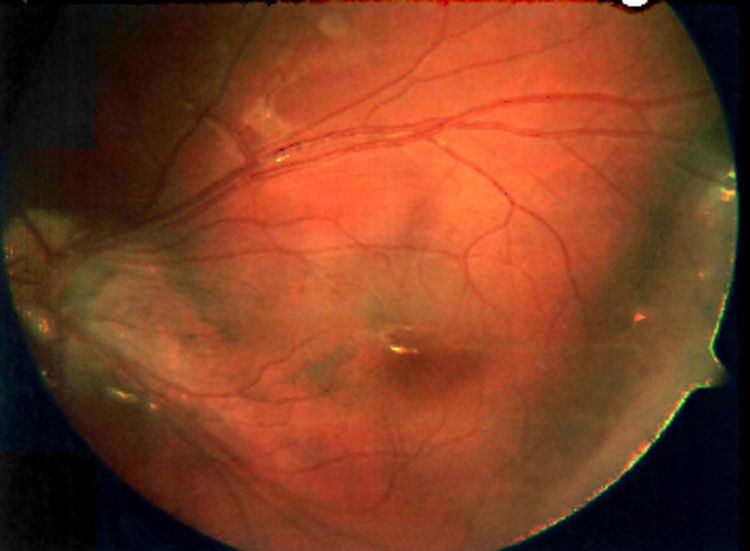
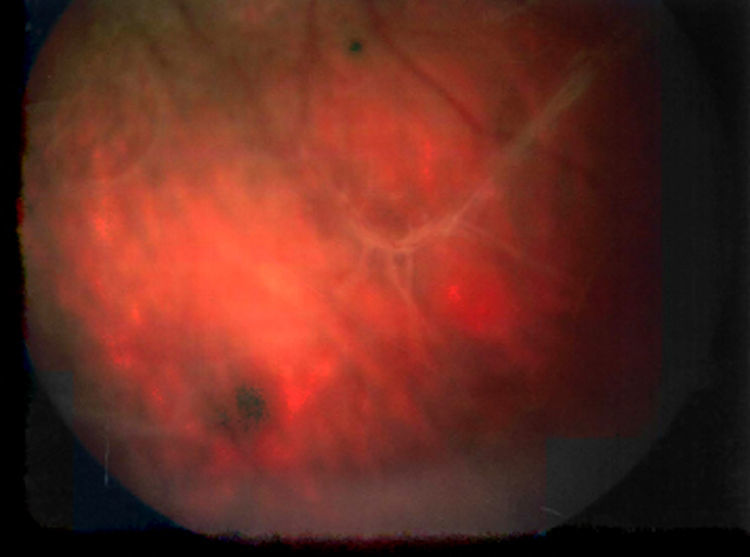
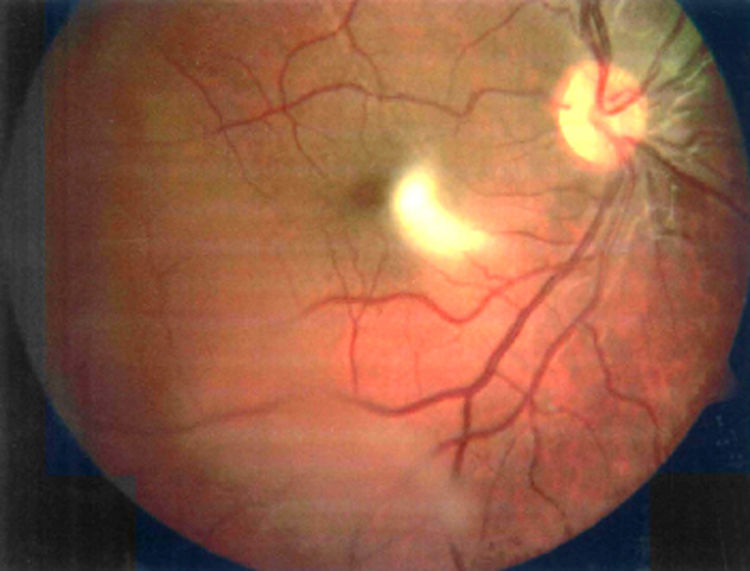
La retinosquisis ligada al cromosoma X es una retinopatía degenerativa de carácter recesivo. Presentamos dos casos clínicos que debutaron con una presentación atípica (estrabismo) en la infancia precoz (lactancia). Ambos niños presentaban velos vítreos en retina periférica. Se encontró una mutación en el gen XLRS1 en ambos casos.
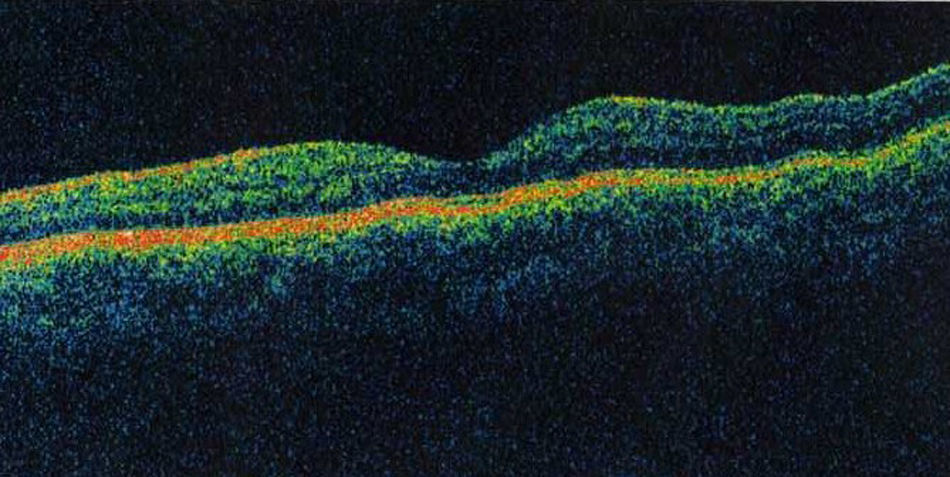
DiscusiónLa retinosquisis ligada al cromosoma X es una de las causas principales de degeneración macular en niños varones. Se caracteriza por una esquisis foveal estrellada, asociada o no a retinosquisis periférica. El diagnóstico clínico puede ser difícil por la alta variabilidad fenotípica del cuadro. Además, el ERG y la OCT pueden ser normales en fases precoces de la enfermedad y de difícil realización en niños pequeños. Consideramos que el cribado para la mutación del gen XLRS1 es útil para evaluar casos con presentación atípica de retinosquisis ligada al cromosoma X y/o en niños en los que otras pruebas complementarias no son realizables.
X linked retinoschisis is a recessively inherited degenerative retinopathy. We report two cases that debuted with an unusual presentation (strabismus) in early childhood (months). Both of them presented with vitreous veils in the retinal periphery. Mutation in the XLRS1 gene was detected in both cases.
DiscussionX linked retinoschisis is one of the leading causes of macular degeneration in male children. Clinical features include a stellate foveal schisis, with or without peripheral retinoschisis. Clinical diagnosis is often difficult because of a high degree of phenotype variability. Furthermore, ERG and OCT may be normal in early stages of the disease. In our opinion, the XLRS1 gene mutation screening provides a powerful clinical tool for evaluating clinically ambiguous cases of X linked retinoschisis.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
