



La diabetes mellitus neonatal es una enfermedad hereditaria monogénica en el 80-90% de los casos. Se presenta de dos formas: una transitoria, por lo general en la primera semana de vida, caracterizada por la asociación de hiperglicemia y restricción del crecimiento intrauterino; y una permanente, que cursa con hiperglicemia severa y cetoacidosis, manifestándose antes de los seis meses de vida. Las formas clínicas dependen de la variación genética, la cual es identificada a través de la secuenciación del exoma completo. El tratamiento se basa en la corrección de la hiperglucemia con insulina, así como el manejo con sulfonilureas a dosis altas, considerándose un tratamiento exitoso, seguro y efectivo para pacientes con diabetes neonatal permanente. Se presenta el caso de un lactante de un mes y 26 días de vida, quien cursa con un cuadro de cetoacidosis diabética, encontrándose en delicadas condiciones generales, al que se le instauró manejo con insulinoterapia con adecuada respuesta de alteraciones metabólicas y neurológicas; se confirmó mutación del gen KCNJ11, que sugiere diabetes mellitus neonatal permanente. En la actualidad el paciente se encuentra en tratamiento con glibenclamida, no presenta deterioro del neurodesarrollo y su crecimiento es satisfactorio. Concluimos que todo paciente menor de seis meses con diagnóstico de diabetes mellitus, independientemente de las manifestaciones clínicas, debe ser estudiado con pruebas genéticas o estudios moleculares con el fin de identificar mutaciones asociadas que determinarán la gravedad de la enfermedad y, por consiguiente, realizar un diagnóstico temprano, tratamiento oportuno y evitar complicaciones o secuelas a largo plazo.
Neonatal diabetes mellitus is a monogenic hereditary disease in 80-90% of cases. It presents in two forms: a transient one, generally in the first week of life, characterized by the association of hyperglycemia and intrauterine growth restriction; and a permanent one that presents with severe hyperglycemia and ketoacidosis that manifests before 6 months of life. Clinical forms depend on genetic variation, which is identified by whole exome sequencing. The right treatment is based on the correction of hyperglycemia with insulin, as well as management with high doses of sulfonylureas. This case shows an infant, 1 month and 26 days old, who presents with diabetic ketoacidosis intially in delicate general condition; who was treated with insulin therapy with an adequate response to the metabolic and neurological alterations; KCNJ11 gene mutation was confirmed, suggesting permanent neonatal diabetes mellitus. Currently the patient is being treated with glibenclamide, there is no deterioration in neurodevelopment and his growth is satisfactory. We conclude that all patients under 6 months of age with a diagnosis of diabetes mellitus, regardless of the clinical manifestations, should be studied with genetic or molecular tests to identify associated mutations that will determine the severity of the disease and therefore allow to make an early diagnosis, timely treatment and avoiding long-term complications.
La diabetes mellitus neonatal (DMN CIE10:P702) es una enfermedad hereditaria monogénica en el 80-90% de los casos1, se presenta de dos formas: una transitoria, que generalmente se manifiesta en la primera semana de vida, caracterizada por la asociación de hiperglicemia con restricción del crecimiento intrauterino2; y una permanente, que cursa con hiperglicemia severa y cetoacidosis, asociada con insulina circulante insuficiente o nula antes de los seis meses de vida (edad media: 7 semanas; rango: desde el nacimiento hasta las 26 semanas) y raramente entre los seis meses y el primer año de edad3. La patogenia de la enfermedad se explica por dos mecanismos principales: malformación del páncreas con alteración de las células secretoras de insulina o función anormal de la célula β pancreática existente. En la actualidad se han reportado alrededor de 23 alteraciones genéticas relacionadas con esta patología1,3. Tiene una incidencia de aproximadamente 1 de cada 90.000-160.000 nacidos vivos y es mayor en países occidentales con altos índices de matrimonios consanguíneos. En cuanto a la prevalencia debida a la mutación del gen KCNJ11, esta es incierta; los estudios informan mutaciones entre el 34-64% de los pacientes con diabetes neonatal permanente (DMNP)4,5.
Para el diagnóstico se debe realizar una prueba genética confirmatoria cuando se presente alguna de las siguientes condiciones:
- 1.
Lactantes con hiperglucemia insulinodependiente.
- 2.
Controles de glicemia superiores 250mg/dl que persisten entre 7 a 21 días de vida, luego de descartar otras posibles etiologías.
- 3.
Hiperglucemia severa (>1.000mg/dl), sin causa aparente, independientemente de la clínica.
- 4.
Lactantes menores de seis meses de edad con diagnóstico de diabetes mellitus, o entre seis meses y un año cuando cursa con manifestaciones extrapancreáticas y/o no hay evidencia de autoinmunidad del páncreas y/o múltiples trastornos autoinmunes o antecedentes familiares inusuales o defectos congénitos asociados5.
Las formas clínicas de la DMN dependen de la variación genética, así como el pronóstico y tratamiento dependen del gen afectado, el cual es identificado a través de la secuenciación de exoma completo6. Los avances en genética han permitido disponer de pruebas más eficientes y completas y son un pilar fundamental debido a los beneficios que brinda a los pacientes afectados, por lo que estarían indicadas en todos los casos de diabetes mellitus diagnosticados antes de los 12 meses de edad1.
El presente reporte de caso describe la evolución clínica de un paciente con DMNP que inicialmente cursa con una cetoacidosis diabética, una presentación clínica poco usual por sus manifestaciones y resultados en la bioquímica sanguínea, aunque con buena respuesta clínica al manejo instaurado, confirmando patología endocrina de base con estudios genéticos.
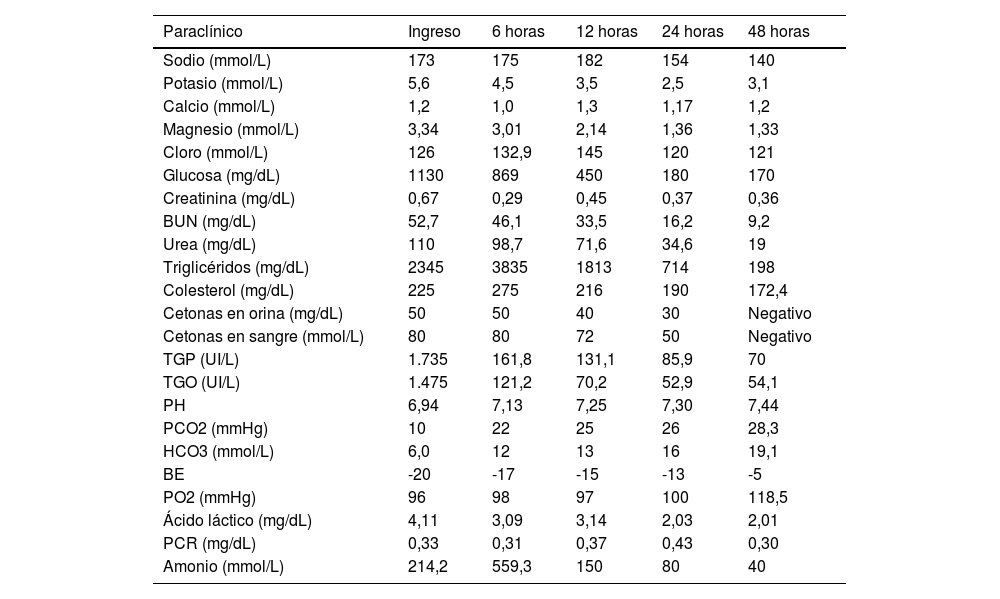
Reporte de casoLactante masculino de 1 mes y 26 días de vida, hijo de madre de 47 años de edad, producto de fertilización in vitro con alto riesgo obstétrico, nace de 39 semanas por cesárea, peso al nacer: 2.950 g, talla al nacer: 48cm, buena adaptación neonatal, egreso con la madre. Antecedentes patológicos: cólicos del lactante y reflujo gastroesofágico. Antecedente familiares: línea materna, tía diabética; línea paterna, bisabuelos diabéticos. Inicio de cuadro clínico caracterizado por alzas térmicas no cuantificadas asociadas a deposiciones líquidas de escasa cantidad, consultan al servicio de Urgencias donde diagnostican infección de vías urinarias confirmada por urocultivo (Escherichia coli multisensible), tratada con antibioticoterapia de amplio espectro (gentamicina–amikacina) por tres días; se egresa con cefalexina oral. Sin embargo, inmediatamente en casa, cursa con múltiples crisis caracterizadas por postura tónica que progresa a movimientos clónicos bilaterales generalizados de corta duración; los padres acuden por consulta externa, donde además refieren aparente intolerancia a fórmulas lácteas, dado por aumento en número de deposiciones y pérdida de peso. El paciente es enviado al servicio de Urgencias por encontrarse hipoactivo, taquipneico, taquicárdico, febril y con signos de deshidratación grado III; ingresa a unidad de Cuidados Intensivos Pediátricos (UCIP) con paraclínicos de ingreso que reportaron (tabla 1) hemograma normal, ionograma con hipernatremia severa, hipercloremia e hipermagnesemia, perfil lipídico con hipercolesterolemia e hipertrigliceridemia; hiperglicemia, cetonemia, cetonuria, función hepática alterada, gases arteriales con acidosis metabólica severa, lactato aumentado, hiperamonemia, función renal alterada, tiempos de coagulación en rango normal, PCR no reactiva.
Evolución de paraclínicos realizados durante estancia en Unidad de Cuidados Intensivos Pediátricos
| Paraclínico | Ingreso | 6 horas | 12 horas | 24 horas | 48 horas |
|---|---|---|---|---|---|
| Sodio (mmol/L) | 173 | 175 | 182 | 154 | 140 |
| Potasio (mmol/L) | 5,6 | 4,5 | 3,5 | 2,5 | 3,1 |
| Calcio (mmol/L) | 1,2 | 1,0 | 1,3 | 1,17 | 1,2 |
| Magnesio (mmol/L) | 3,34 | 3,01 | 2,14 | 1,36 | 1,33 |
| Cloro (mmol/L) | 126 | 132,9 | 145 | 120 | 121 |
| Glucosa (mg/dL) | 1130 | 869 | 450 | 180 | 170 |
| Creatinina (mg/dL) | 0,67 | 0,29 | 0,45 | 0,37 | 0,36 |
| BUN (mg/dL) | 52,7 | 46,1 | 33,5 | 16,2 | 9,2 |
| Urea (mg/dL) | 110 | 98,7 | 71,6 | 34,6 | 19 |
| Triglicéridos (mg/dL) | 2345 | 3835 | 1813 | 714 | 198 |
| Colesterol (mg/dL) | 225 | 275 | 216 | 190 | 172,4 |
| Cetonas en orina (mg/dL) | 50 | 50 | 40 | 30 | Negativo |
| Cetonas en sangre (mmol/L) | 80 | 80 | 72 | 50 | Negativo |
| TGP (UI/L) | 1.735 | 161,8 | 131,1 | 85,9 | 70 |
| TGO (UI/L) | 1.475 | 121,2 | 70,2 | 52,9 | 54,1 |
| PH | 6,94 | 7,13 | 7,25 | 7,30 | 7,44 |
| PCO2 (mmHg) | 10 | 22 | 25 | 26 | 28,3 |
| HCO3 (mmol/L) | 6,0 | 12 | 13 | 16 | 19,1 |
| BE | -20 | -17 | -15 | -13 | -5 |
| PO2 (mmHg) | 96 | 98 | 97 | 100 | 118,5 |
| Ácido láctico (mg/dL) | 4,11 | 3,09 | 3,14 | 2,03 | 2,01 |
| PCR (mg/dL) | 0,33 | 0,31 | 0,37 | 0,43 | 0,30 |
| Amonio (mmol/L) | 214,2 | 559,3 | 150 | 80 | 40 |
BUN: nitrógeno ureico en sangre; TGP: transaminasa glutámico pirúvica; TGO: transaminasa glutámica oxalacética; PCO2: presión parcial dióxido de carbono; HCO3: bicarbonato; PO2: presión parcial de oxígeno; PCR: proteína C reactiva.
El manejo inicial fue para la corrección de la cetoacidosis y los trastornos hidroelectrolíticos, se indicó fluidoterapia e infusión de insulina cristalina endovenosa a dosis inicial de 0,01 UI por kg/hora con el objetivo de mantener la glicemia entre 150 a 200mg/dl.
Paciente con mejoría de alteraciones metabólicas a las 24 horas, es valorado por endocrinología pediátrica, que inicia esquema subcutáneo con insulina de acción prolongada (detemir) a dosis de 0,11 UI/kg/día cada 24 horas; durante su estancia en UCIP cursa con niveles de glucometrías fluctuantes con tendencia a la hipoglicemia, por lo que se indicó bomba de infusión de insulina; alrededor del día 40, paciente con mejoría de controles de glicemia, por lo que se inicia manejo con sulfonilureas tipo glibenclamida a dosis de 0,1mg con adecuada respuesta clínica que permite la suspensión definitiva de insulina a los 10 días.
Es importante resaltar que el abordaje diagnóstico multidisciplinario incluyó evaluación por Genética y Neurología, considerando patologías como error innato del metabolismo, aciduria orgánica, glucogenosis y encefalopatía metabólica, por lo que se solicitaron estudios de extensión como tomografía axial computarizada (TC) de cráneo simple, resonancia magnética (RM) de cerebro con espectroscopía y punción lumbar para establecer vía metabólica asociada, con resultados dentro de la normalidad; Endocrinología Pediátrica sugiere estudios moleculares (genes ABCC8 y KCJN11) para confirmar diagnóstico, establecer pronóstico y tratamiento; la secuenciación del exoma completo identifica estado de heterocigosis la variante patogénica c.175G>A; p. Val59Met en el gen KCNJ11 relacionada con susceptibilidad a DMNP, con o sin características neurológicas, con un patrón de herencia autosómico dominante. Paciente egresa en tratamiento con glibenclamida, sin efectos adversos posterior a los tres meses, no presenta deterioro del neurodesarrollo y su crecimiento es satisfactorio.
DiscusiónLa DMNP está asociada a una alteración genética que afecta el canal de potasio sensible ATP, se han encontrado mutaciones asociadas al gen KCNJ11 que se atribuyen a la forma permanente en el 30-66% de los casos, con herencia autosómica dominante. Este gen codifica la subunidad kir6.2, localizada en el canal de potasio en las células beta pancreáticas; tras la captación de glucosa se produce un incremento de ATP/ADP a nivel intracelular, provocando el cierre de los canales ATP de potasio, ocasionado la despolarización de la membrana celular con posterior apertura de los canales de calcio dependientes de voltaje, que conlleva a la entrada del mismo a nivel intracelular liberando la insulina; en los pacientes que presentan alteración a nivel del gen KCNJ11 no hay cierre de los canales de potasio y, por consiguiente, no hay ingreso de calcio para la liberación de insulina6.
El fenotipo de esta patología está asociado a la severidad de la mutación y expresión del gen. El principal síntoma es la hiperglucemia marcada, bajo peso al nacer, lo que denota la ausencia de la liberación de insulina; a nivel del sistema nervioso se presenta retraso en el neurodesarrollo, alteración del patrón del sueño, trastorno del aprendizaje y epilepsia como complicaciones tardías; por lo anterior, es imperativo realizar diagnósticos neuropsiquiátricos. Se han descrito casos de pacientes con la mutación y desarrollo de este tipo de trastornos; así mismo, se debe tener en cuenta la tríada clásica del síndrome de DEND (diabetes neonatal, retraso del desarrollo neurológico, epilepsia) asociada a la mutación en el gen KCNJ117.
El diagnóstico genético es importante debido a que determina la gravedad de la enfermedad y repercute en el manejo terapéutico. De León1, Gole3, Hattersley8, entre otros, manifiestan que estos pacientes podrían recibir solo tratamiento con sulfonilureas y no quedar insulinodependientes, como lo menciona Park9, quien sugiere que la bomba de insulina es un modo seguro y eficaz para tratar la DMN y su utilización temprana puede acortar la duración de las estancias hospitalarias, pensando en lo difícil que es dosificar la insulina y al estrecho margen terapéutico entre hipoglucemia e hiperglucemia que podrían repercutir en el desarrollo neurológico del lactante. Tonini10 propone como opción en los pacientes con DMNP y mutación KCNJ11 una transición de la insulina a la sulfonilurea, considerando que el uso de bombas con sistema automatizados de administración de insulina (AID) y monitorización remota de pacientes, permite un buen control glicémico y posibilita una transición exitosa con sulfonilurea, minimizando los picos de hiperglicemia, hipoglicemia y, sobre todo, mejor calidad de vida al paciente y familiares11; teniendo en cuenta que el 90% de los pacientes toleran con éxito el cambio de la insulina a las sulfonilureas orales a dosis altas, actualmente es un tratamiento apropiado para pacientes con DMNP; esta terapia es segura y altamente efectiva, manteniendo un excelente control glucémico durante al menos 10 años; como consecuencia un gran porcentaje de pacientes podrían suspender el manejo con insulina12,13.
Según el reporte descrito por Sánchez14, es importante prevenir las complicaciones agudas secundarias, tales como cetoacidosis diabética y la hipoglucemia, así como las tardías anteriormente descritas, instaurando tratamiento agresivo, control frecuente de las concentraciones de glucosa en sangre al menos cuatro veces al día y evaluaciones periódicas del desarrollo. Posterior a 10 años, se indican pruebas de detección anuales para descartar complicaciones crónicas de la diabetes mellitus, incluido un uroanálisis para detectar microalbuminuria y un examen oftalmológico para descartar retinopatía.
Cabe resaltar que dentro de la literatura existente encontramos diferentes formas de presentación de acuerdo a la variante reportada en los estudios genéticos, como lo es el caso descrito por Gole3 en el año 2018, sobre un paciente de origen paquistaní de 22 días de vida, quien cursa con signos de dificultad respiratoria y deshidratación severa, el estudio diagnóstico reveló hiperglucemia con cetoacidosis grave (glucosa: 907mg/dl, pH de los gases en sangre: 6,84, HCO3: 6 mmol/l), que se manejó con líquidos intravenosos (IV) y administración de insulina IV, se realiza prueba genética que identifica mutación del gen KCNJ11 (p.P254Q), que conduce a diabetes mellitus neonatal transitoria, que en este caso desaparece a los 10 meses de vida.
También encontramos el reportado por Poon15 en el 2022, quien describe un paciente que cursó con diabetes mellitus neonatal y cetoacidosis diabética a los 10 meses de edad con autoanticuerpos pancreáticos indetectables a lo largo de su vida, lo que conllevó a realizar pruebas genéticas a los 13 años de edad, encontrándose una variante heterocigótica C42R, en el gen KCNJ1115.
El presente reporte de caso describe el diagnóstico clínico y genético de la DMNP por mutación del gen KCNJ11 con una variante patogénica no descrita anteriormente, que se manifiesta como una cetoacidosis diabética con control satisfactorio al iniciar manejo con insulina y posterior seguimiento con sulfonilurea. Una fortaleza importante es que se abarcaron todos los diagnósticos diferenciales posibles para este grupo etáreo, lo cual demuestra un manejo integral y direccionado para la patología; sin embargo, una de las más grandes limitaciones fue la demora en la realización y reporte de los estudios genéticos.
ConclusiónConcluimos que todo paciente menor de seis meses con diagnóstico de DMN, independientemente de las manifestaciones clínicas, debe ser estudiado con pruebas genéticas o estudios moleculares, que permitan identificar mutaciones asociadas que determinarán la gravedad de la enfermedad, y por consiguiente, nos orientarán hacia un diagnóstico temprano, tratamiento oportuno proporcionando terapias que puedan administrarse por vía oral, así como evitar complicaciones o secuelas a largo plazo.
FinanciaciónEste reporte de caso no recibió financiación económica para su realización.
Conflicto de interesesNo se declaran conflictos de intereses para este artículo.
Agradecemos a la Dra. Leticia Martínez Ariza, endocrinóloga pediatra, por sus valiosos aportes en cuanto al diagnóstico, tratamiento y seguimiento del paciente.
