




Neurodegenerative diseases are reliant on neurons which demand high energy for its functioning, which depends on systems like mitochondrial oxidative phosphorylation (OXPHOS). This energy generated by mitochondria occurs through different respiratory complexes ranging from complex I to IV. Complexes are also known as NADH, including complex I because it is the first enzyme and largest chain of the given respiratory chain; however, complex 1 is called the ubiquinone oxidoreductase. Majority of neurodegenerative cases demonstrate irresistible evidence of plenty of mitochondrial subtleties that are impaired, as inhibition of complex 1 could lead to excessive reactive oxygen species (ROS) production.
DevelopmentIn this review we try to discuss on how epigenetic modifications could cause a drift in smooth functioning of mitochondria and its function leading to neurodegenerative diseases like Leber's hereditary optic neuropathy (LHON). The reference articles were collected from NCBI-Pubmed, Scopus and Web of science. This review tries to deliver a summarizing overview related to Complex 1 mediated mitochondrial dysfunction and studies done so far in this aspect, along with the epigenetic modifications.
ConclusionIn this review we discussed the different roles of mitochondria and its related dysfunction in the pathogenesis of neurodegenerative diseases.
Las neuronas necesitan grandes cantidades de energía para su funcionamiento y dependen de sistemas como la fosforilación oxidativa mitocondrial. Dicha energía se genera en las mitocondrias a través de diferentes complejos respiratorios (complejos I-IV). El complejo I, también conocido como nicotinamida adenina dinucleótido (NADH por sus siglas en inglés) o ubiquinona oxidorreductasa, es la primera enzima y el mayor complejo de la cadena respiratoria. La mayoría de enfermedades neurodegenerativas se asocian de forma inequívoca a una gran variedad de alteraciones mitocondriales, como la inhibición del complejo I, que podría desencadenar una sobreproducción de especies reactivas de oxígeno.
DesarrolloEl objetivo de nuestra revisión es analizar cómo las modificaciones epigenéticas pueden alterar el funcionamiento normal de las mitocondrias, lo que puede a su vez favorecer el desarrollo de enfermedades neurodegenerativas como la neuropatía óptica hereditaria de Leber. Hemos usado las bases de datos PubMed, Scopus y Web of Science para identificar artículos relacionados. Esta revisión presenta un resumen de la evidencia disponible hasta la fecha sobre la disfunción del complejo I mitocondrial y las modificaciones epigenéticas identificadas.
ConclusiónAnalizamos el papel de las mitocondrias y las disfunciones mitocondriales en la patogénesis de las enfermedades neurodegenerativas.
Mitochondrial diseases are inherited genetically which occurs when mitochondria is unable to proceed oxidative phosphorylation. This happens due to mutations in mitochondrial genes and nuclear genes which encodes structural mitochondrial proteins. Mitochondrial DNA mutations are transmitted by maternal inheritance and nuclear DNA mutations may follow an autosomal dominant, autosomal recessive or X-linked inheritance pattern. Mitochondrial diseases prevalence is estimated around 1 in 5000 in adults.1 Deficiency of complex 1 due to pathogenic mutations leads to various mitochondrial diseases. Various nuclear gene as well as mitochondrial gene mutations have been identified which has been associated with mitochondrial complex 1 deficiency. Mutations in genes which codes for structural subunit and assembly factors of complex 1 is referred as Primary complex 1 defects.2 Factors such as mitochondrial dysfunction, oxidative stress and apoptosis have an important role in various ocular diseases whereas oxidative stress and mutations in mitochondrial protein encoding genes are associated with several neurodegenerative diseases.3 Epigenetic regulation among nuclear geneswhich encodes for complex 1 subunitswould make evidentchanges in expression of complex 1 subunit. This changes in mitochondrial protein level would lead to defective oxidative phosphorylation and energy depletion in cells.
Focusing on the range of genomic mechanism which not only relies on the sequence of DNA but also other factors is epigenetics. It is important to integrate cues from the environment in order to understand the development of regulated genetic complication and control homeostasis. There are various studies carried out in epigenetics against embryology, cellular differentiation and aging. In the last decade there are many pioneering studies focusing on epigenetics that include acetylation, methylation, ubiquitylation and phosphorylation which are natural occurring processes that take place in every organism, and changes in these can cause unwanted effects on health leading to various diseases/disorders.4 Researches in epigenetic regulation of nuclear encodedmitochondrial genes will lead to better understanding of various mitochondrial diseases and increases the chances to find out better therapeutics in future.
Mitochondria and oxidative phosphorylationOxidative phosphorylation consists of five enzyme complexes which are embedded in the inner mitochondrial membrane.5 Mitochondria is the only one organelle in eukaryotic cell which have their own genome separately. The mtDNA is located in the matrix and each mitochondria contain several identical copies of mtDNA.6 Other than the energy production, mitochondria have several unique function in control of cell cycle, regulating cell signaling, maintaining cell death and other cellular metabolism.7 Oxidative phosphorylation is a complex process and OXPHOS system that requires a large group of mitochondrial as well as nuclear encoded proteins to function accurately.8 OXPHOS system includes complex I (CI or NADH:ubiquinone oxidoreductase), complex II (CII or succinate:ubiquinone oxidoreductase), complex III (CIII or ubiquinol:cytochrome-c oxidoreductase), complex IV (CIV or cytochrome-c oxidase) and Complex V (FOF1-ATP-synthase). The flow of electrons from NADH/FADH2 to O2 through this subunits located in the mitochondrial inner membrane leads to the pumping of protons out of the mitochondrial matrix and its generate ATP.9
Role of complex 1 in oxidative phosphorylationOXPHOS subunit Complex I (NADH:ubiquinone oxidoreductase) is the largest subunit and C1 is boot shaped structure of ~1 MDa and being composed of the hydrophobic membrane arm and the hydrophilic matrix arm perpendicular to each other. The matrix arm consists mostly of the nuclear-encoded subunits, FMN, three iron–sulfur clusters and site for internal ubiquinone. The membrane arm has all mtDNA-encoded subunits, one iron–sulfur cluster and catalytic site for the substrate ubiquinone.10 Central C1 subunits arranged as three functional modules: 1) N module, the electron input module that oxidizes NADH 2) Q module that reduces ubiquinone and 3) P module that translocate proton across the membrane.11 N module comprising NDUFV1, NDUFV2, NDUFS1 subunits, in which electron entering to N module through FMN from NADH. FMN is non-covalently bound to NDUFV1 and it transfers the electron to Fe–S clusters N3, N1b, N4, and N5.12 The Q module is composed of NDUFS2, NDUFS3, NDUFS8, and NDUFS7 subunits and it accepts electrons from the iron–sulfur clusters of the N module and transfers the electrons through iron–sulfur clusters N6a, N6b and N2 to ubiquinone.13 Study has been reported about complex 1 deficiency in drosophila model which is caused by homoplasmic mutation in ND2 gene. This mutant model exhibits phenotypes that resemble symptoms of mitochondrial disease, short lifespan, progressive neurodegeneration, altered mitochondrial membrane potential and low level ATP production.14 P module is membrane embedded and it mediates proton pumping. P module comprising the seven mitochondrial DNA-encoded CI subunits (ND1, ND2, ND3, ND4, ND4L, ND5, ND6), ND1 at the base of the Q-module, followed by ND3, ND6 and ND4L, and the antiporter-like subunits ND2, ND4 and ND5.11,15 Majority of the complex 1 subunit proteins for assembly and accessory factors are encoded by nuclear genes, those proteins are synthesized in cytosol and imported into the mitochondria through translocation machineries.16
There are studies which show that biosynthesis of complex 1 is a complicated process and many assembly factors are involved in each step of the process.17 Many studies have been conducted to understand the different stages of complex 1 assembly process and its integration into the respiratory chain super complexes.18 Recent study proving that modules of complex 1 were found to form five different subassemblies, comprising all 14 central and 18 accessory subunits corresponding to almost 80% of the total mass of fully assembled complex I.18 The N module core subunits NDUFV1, NDUFV2 and NDUFS1 and accessory subunits NDUFV3, NDUFS4, NDUFS6 and NDUFA12 are integrated at last to form complex 1 functional holo-enzyme.19 Accessory subunits form a scaffold around the core subunit to prevent ROS generation and for the protection against oxidative damage by shielding redox groups from reaction with oxygen, in binding of the CI to the membrane, and in the stabilization of the enzyme. Also, more specific functions in the regulation of activity or the assembly of other subunits into the holocomplex have been implied.20 The Complex I assembly factors NDUFAF1, ACAD9, ECSIT, TMEM126B, and TIMMDC1 form the MCIA complex, which was identified in human osteosarcoma 143B cells by complexome profiling.21
Complex 1 mediated ROS production and oxidative stressOxidative stress is the imbalance between reactive oxygen species (ROS)/reactive nitrogen species (RNS) generation and the biological antioxidant defense system. The accumulation of ROS in cells and changes in functional level of macromolecules leads to damage of tissue to organ and contributes to pathogenensis of many diseases.22 ROS production can happen in mitochondria in two different ways: (1) In results of complex 1 inhibition, the electrons are backed up within the Fe–S cluster of complex 1. (2) Reverse electron transfer, where electrons are going backward to complex 1 from complex II via ubiquinone which generate superoxide from semi-ubiquinone at the quinine-binding site.23 Mitochondrial inner membrane is the major site of ROS production and mitochondrial DNA (mtDNA) are more prompt for ROS damage. mtDNA damage is more substantial and persist longer than mutations in nuclear DNA.24 mtDNA is more susceptible for oxidative stress than nDNA because of its close proximity to the inner mitochondrial membrane: major production of ROS site as well as mtDNA is not protected with histone or any other associated proteins and intronless regions, all these factors enhance the oxidative modification in the genome. Effect of mitochondrial damage reflects on the mtDNA as well as mtRNA transcrits reduced level and alterations in mitochondrial function.25 ROS also attacks structural and enzymatic proteins through the oxidation of aminoacids, prosthetic group, formation of cross links and proteolysis. Inhibition of key proteins can lead to serious consequences in the metabolic pathways. mtDNA can be displaced to intra- and extracellular compartments in response to various stress conditions. Displacement of mtDNA from mitochondria is still unclear and studies are indicating that through the generation and release of extracellular vesicles mtDNA transfer to extracellular compartment is possible.26,27
Studies have illustrated that several mitochondrial diseases are associated with ROS mediated mtDNA mutation. In MELAS, hydroxyl radical damage to mtDNA and it can accelerate the mitochondrial genotype associated with the disease.28 In MELAS and MERRF patients, intracellular ROS level and oxidative damage to macromolecules were increased in skin fibroblast cells.29 ROS associated telomerase shortening have been observed in patients with MELAS-related mitochondriopathy and patient with LHON.30 In Leber's hereditary optic neuropathy (LHON), all associated point mutations such as ND1, ND4 and ND6 are enhancing ROS production.31 Increased or decreased ROS production leads to oxidative stress condition in cells and it induces mitochondrial DNA mutations, damage the mitochondrial respiratory chain, amend membrane permeability and it has impact on Ca2+ homeostasis and mitochondrial defense systems.32 As a consequence of LHON point mutation, oxidative stress is responsible for cellular damage and activates apoptosis in RGCs.33 Oxidative stress cause DNA damage and level of DNA damage marker (8OH2'dG) has been shown raised in LHON patient leukocyte.34 Human osteosarcoma cybrid cells with LHON mutations were also showed reduced GSH activity.35 In animal model study showed ATP production remains same while oxidative stress remains as high indicating the role of ROS as a source of RGC degeration.36
Diseases associated with mutations in mitochondrial complex 1 dysfunctionAssembly defects in complex 1 lead to improper functioning of electron transport chain and severe disease conditions. Various mutations in mitochondrial and nuclear genes associated with complex 1 cause most of the mitochondrial diseases.37 Almost 25 genes have been identified for complex 1 deficiency in which 6 genes are nuclear encoded accessory protein genes and 19 mitochondrial genes codes for complex 1 subunits. Proper assembly of subunits and accessory proteins are required for the stability and functioning of complex 1.38 Pathogenic mutations have been identified in mitochondrial genes for Leigh syndrome, multisystem disorders such as MELAS (mitochondrial encephalopathy with lactic acidosis and stroke like episodes), or MELAS/LHON overlap syndromes,39 Leber's Hereditary Optic Neuropathy (LHON), and MERRF (myoclonic epilepsy and ragged red fibers), Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes.
Syndrome resembling strokes are associated with severe complex I deficiency caused by mitochondrial DNA (mtDNA) mutations.12 Among mitochondrial disorders, MELAS syndrome is considered as one of the most common maternally inherited diseases and the pathogenicity of MELAS is still not well understood.40 Study using MELAS neuronal cybrid cells reported a homoplasmic m.3243A > G mutation which is a metabolic switch towards glycolysis with the production of lactic acid. It shows severe defects in respiratory chain activity and complex I disassembly with an accumulation of assembly intermediates. Studies showed that Ketone Bodies (KB) exposure during 4 weeks associated with glucose deprivation significantly restored complex I stability and activity, increased ATP synthesis and reduced NADH/NAD+ ratio, mtDNA copy number were significantly increased with KB. KB may constitute an alternative and promising therapy for MELAS syndrome, and could be beneficial for other mitochondrial diseases caused by complex I deficiency.41
Mutations in nuclear encoded structural subunit genes and in assembly factor genes are associated with severe clinical presentations like Leigh syndrome, Leigh like syndrome, leukoencephalopathy with macrocephaly, leukoencephalopathy, lethal infantile mitochondrial disease, cardiomyopathy and encephalopathy, unspecified encephalomyopathy, progressive cavitating leukoencephalopathy, hypertrophic cardiomyopathy and encephalomyopathy are associated with mutations in nuclear encoded complex 1subunits.42,43 In 1998, first study about mutation in nDNA encoded complex 1 subunit was reported and the study was on heterozygous mutation in NDUFS8 gene in a patient with Leigh syndrome with complex 1 enzyme deficiency in muscle tissue and cultured skin fibroblast.44 Leigh syndrome is the most common clinical presentation of mitochondrial diseases in children and mutation causing Leigh syndrome have been detected in almost all complex 1 gene including both nuclear and mitochondrial genes.45 The emerging use of whole exome sequencing (WES) has become an essential tool in identification of the heterogeneous molecular background of complex I deficiency and discovery of novel disease genes underlying this condition.46
Oxidative stress and complex 1 associated neurodegenerative diseasesBrain tissue has high energy demand and central nervous system functions strongly depending on efficient mitochondrial function. Mutations in mitochondrial genome, alterations in mitochondrial dynamics, ROS generation, pathologic protein aggregations and environmental factors may alter energy metabolism and it leads to neurodegenerative diseases.73 Oxidative stress mediate mitochondrial alterations which might be a major reason for neurodegeneration.74 Neurodegenerative diseases are a heterogeneous group of disorders characterized by gradually progressive, selective loss of neuronal systems which are physiologically or anatomically related. The most common chronic neurodegenerative diseases associated with aging and aggregation of misfolded proteins are AD, PD, HD and ALS. Many studies have proved that complex 1 activity significantly decrease in cortex, cerebellum and brainstem mitochondria from aged sample group compared to young control group.75,76 Neuronal damage occurs as the effect of imbalance of ATP production, membrane potential and calcium uptake, in neurodegenerative processes, increased ROS leads to all these imbalance conditions. In 1990, first study has been reported about the role of mitochondria in neurodegenerative process associated with Parkinson's disease where complex I deficiency was observed in substantia nigra and platelet mitochondria of patients.77 Mitochondrial complex 1 activity is reduced in the substantia nigra of PD patients and study using complex I inhibitors such as rotenone, MPTP and pesticides cause neurological changes similar to PD.78 Partial inhibition of complex I in nerve terminals is sufficient for mitochondria to generate more ROS. H2O2 plays a major role in inhibiting complex I as well as ketoglutarate dehydrogenase which results in higher ROS production in dopaminergic neurons leading to excessive oxidative stress and ATP deficiency that eventually will result in cell death in the nigro-striatal pathway.79 In a PD related study demonstrated that complex 1 inhibition increased free radicals and reduced proteasomal activity via adenosine triphosphate (ATP) depletion and it increases neuronal vulnerability.80 Combinational effect of decreased complex 1 activity, reduced cellular ATP levels, increased ROS levels lead to reduced ATP dependent axonal transport, reduced proteosomal degradation and cytoplasmic accumulation of the tau protein. Hyperphosphorylated tau may be one of the factor that contribute to the apoptosis process and its sufficient to cause neurodegeneration.81 Accumulation of mutant aggregate-prone proteins induced by proteasomal inhibition in neurodegenerative diseases results in impairment of the mitochondrial respiratory chain complexes activity.82
In AD, membrane-associated oxidative stress, increased free radical production and perturbed Ca2+ homeostasis have been observed and also COX activity is reduced and neurons exhibit mitochondrial damage and apoptosis.83 Changes in oxidative metabolism in AD showed as a result of changes in gene expression of nuclear and mitochondrial genes encoding for complex 1 enzyme. Decreased complex 1 activity as the result of down regulation of mitochondrial genes have been found in AD brain specimens.84,85 Decreased gene expression of ND4 subunit of complex 1 also has been reported in the temporal cortex of AD patients.86 Appearance of h Aβ aggregation and Tau pathologies correlate with mitochondrial dysfunctions in neurons in AD.Elevated Ca2+ and ROS levels during mitochondrial dysfunction both contribute to the accumulation of pTau aggregates. In 2007, Melov et al. showed that mitochondrial SOD2 deficiency can result in pTau aggregation in mice, a symptom that is reversible by the administration of antioxidants.87
Other neurological disorders like bipolar disorder and schizophrenia are also linked to mitochondrial dysfunction and oxidative stress. It still remains unclear whether mitochondrial dysfunction is specifically associated with complex I impairment or associated with increased oxidative damage and, if so, whether this relationship is specific to bipolar disorder.88 Levels of NDUFS7 and complex I activity were decreased significantly in patients with bipolar disorder but were unchanged in those with depression and schizophrenia compared with controls88 (Table 1).
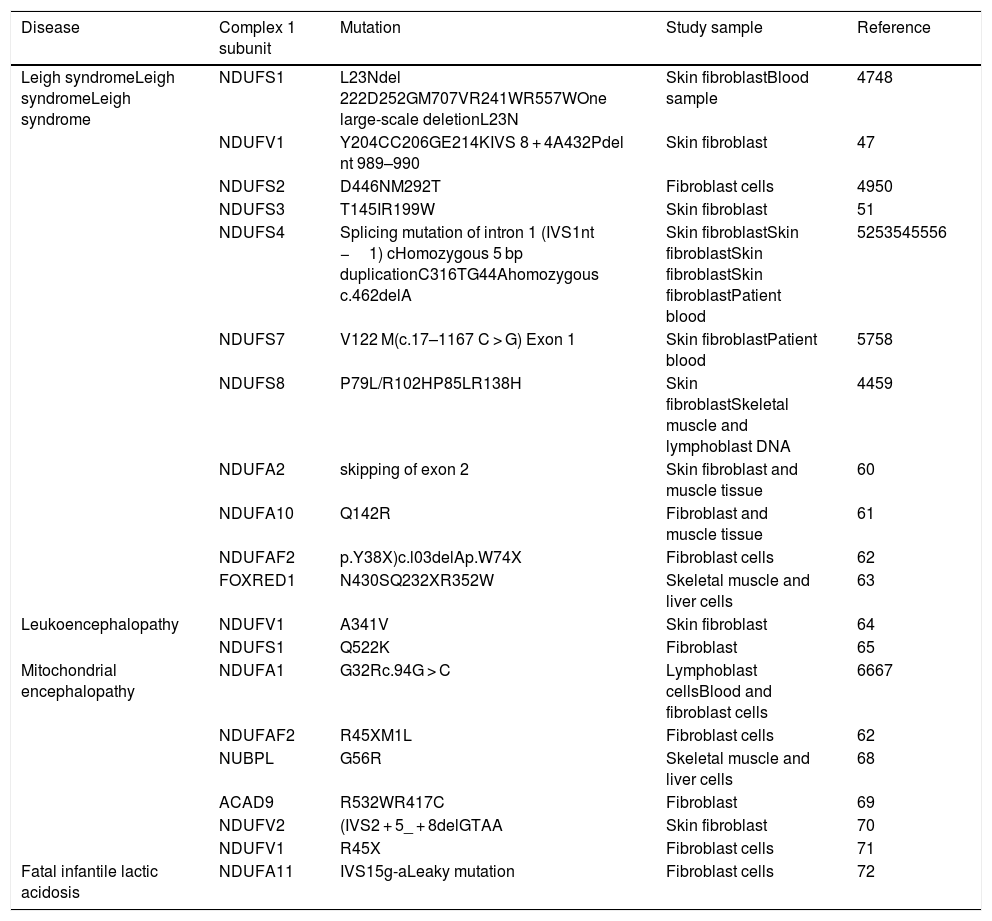
List of diseases with mutations in nuclear encoding genes for mitochondrial complex 1 subunits and assembly factors.
| Disease | Complex 1 subunit | Mutation | Study sample | Reference |
|---|---|---|---|---|
| Leigh syndromeLeigh syndromeLeigh syndrome | NDUFS1 | L23Ndel 222D252GM707VR241WR557WOne large-scale deletionL23N | Skin fibroblastBlood sample | 4748 |
| NDUFV1 | Y204CC206GE214KIVS 8 + 4A432Pdel nt 989–990 | Skin fibroblast | 47 | |
| NDUFS2 | D446NM292T | Fibroblast cells | 4950 | |
| NDUFS3 | T145IR199W | Skin fibroblast | 51 | |
| NDUFS4 | Splicing mutation of intron 1 (IVS1nt −1) cHomozygous 5 bp duplicationC316TG44Ahomozygous c.462delA | Skin fibroblastSkin fibroblastSkin fibroblastSkin fibroblastPatient blood | 5253545556 | |
| NDUFS7 | V122 M(c.17–1167 C > G) Exon 1 | Skin fibroblastPatient blood | 5758 | |
| NDUFS8 | P79L/R102HP85LR138H | Skin fibroblastSkeletal muscle and lymphoblast DNA | 4459 | |
| NDUFA2 | skipping of exon 2 | Skin fibroblast and muscle tissue | 60 | |
| NDUFA10 | Q142R | Fibroblast and muscle tissue | 61 | |
| NDUFAF2 | p.Y38X)c.l03delAp.W74X | Fibroblast cells | 62 | |
| FOXRED1 | N430SQ232XR352W | Skeletal muscle and liver cells | 63 | |
| Leukoencephalopathy | NDUFV1 | A341V | Skin fibroblast | 64 |
| NDUFS1 | Q522K | Fibroblast | 65 | |
| Mitochondrial encephalopathy | NDUFA1 | G32Rc.94G > C | Lymphoblast cellsBlood and fibroblast cells | 6667 |
| NDUFAF2 | R45XM1L | Fibroblast cells | 62 | |
| NUBPL | G56R | Skeletal muscle and liver cells | 68 | |
| ACAD9 | R532WR417C | Fibroblast | 69 | |
| NDUFV2 | (IVS2 + 5_ + 8delGTAA | Skin fibroblast | 70 | |
| NDUFV1 | R45X | Fibroblast cells | 71 | |
| Fatal infantile lactic acidosis | NDUFA11 | IVS15g-aLeaky mutation | Fibroblast cells | 72 |
Ocular diseases also involved in mitochondrial diseases and ocular changes are noticeable feature in various mitochondrial diseases. Eye is high energy demanding organ and mitochondrial DNA damage as the result of oxidative stress studied in pathogenesis of various opathalmologic diseases such as diabetic retinopathy (DR), age-related macular degeneration (AMD) and glaucoma.89 First study in DR demonstrate the overall mtDNA damage in the retina in animal models and shows that this damage continues even after hyperglycemia is reversed, suggesting a strong role for mtDNA damage in the development, and also in the metabolic memory associated with the progression of DR.90 Mitochondrial abnormality in ophthalmic disease associated with complex 1 dysfunction in LHON leads to optic nerve damage specifically involving retinal ganglion cells.
Leber's Hereditory Optic Neuropathy (LHON) is a maternally inherited mitochondrial disorder which leads to bilateral vision loss by degeneration of retinal ganglion cells. Point mutations in the mtDNA which code for NADH dehydrogenase subunit of Complex 1 in the ETC causes the progressive vision loss.91 The three common primary mitochondrial mutations are m.11778G4A in MT-ND4 gene, m.3460G4A in MT-ND1gene and m.14484T4C in MT-ND6 gene. The prevalence for the disease is estimated to 1:3000 and LHON begins generally in the mean age onset between 18 and 35, it happens around 50% in male and 10%–50% in females.92 In the earlier stage of the disease, acute or subacute, painless vision loss in one eye and gradually within few weeks it will affect the second eye also. Majority of patient will remain blind and it affect the overall quality of their life.93 Sometimes LHON misdiagnosed as optic neuritis at early stage due to acuity of onset, age at presentation.94 Recently one study has been reported, a case with painful unilateral optic neuritis pave the way for the onset of LHON. It is not clear whether any mechanism are commonly sharing with these diseases or optic neuritis acted as a activating factor for LHON disease condition.95 Both the incomplete penetrance and the male predominance in LHON are not fully explained yet.
There is no proper treatment to prevent the vision loss caused by LHON till now but medicines are available to promote ATP and decrease the oxidative stress.96,97 Recent study gives evidence that use of idebenone may be beneficial for LHON treatment and it has influential effect on visual acuity (VA) and visual evoked potential (VEP) in LHON patients.98 LHON is considered as a multifactorial disease with complex genetic and environmental interaction. Apart from the mitochondrial mutations, epigenetic factors play an important role in the disease pentrance and expression of the disease. On the basis of clinical studies, several environmental factors have been proposed to prompt LHON. Mitochondrial genetic variants associated with various diseases have been given in Table 2.
Mitochondrial genetic variants associated with various diseases.
| Disease | Mitochondrial genetic variant | |
|---|---|---|
| Leigh syndrome (LS) | MT-ND1 | m.4142 G > T |
| MT-ND3 | m.10158 T > Cm.10191 T > Cm.10197G > A | |
| MT-ND4 | m.11777 C > Am.11778 G > A | |
| MT-ND5 | m.13513G > Am.12706 T > Cm.13094 T > Cm.12706 T > C | |
| MT-ND6 | m.14487T > Cm.14453 G > Am.14459 G > Am.14600G > A | |
| MT-ATP6 | m.8993 T > Gm.8993 T > Cm.9176 T > Gm.9176 T > Cm.9185 T > Cm.9191 T > C | |
| MT-TK | m.8363A > G | |
| MT-TW | m.5537insT | |
| MT-CO3 | m.9537insC | |
| MELAS | MT-ND1 | m.3697G > Am.3946G > Am.3949 T > Cm.3308 T > Cm.3376G > Am.3481G > Am.3959G > Am.3995A > G |
| MT-ND4 | m.12015 T > Cm.11084A > Gm.11470A > C | |
| MT-ND5 | m.12770A > Gm.13046 T > Cm.13063G > A | |
| MT-ND6 | m.14453G > A | |
| MT-TL1 | m.3243A > Gm.3271 T > C | |
| MT-CO3 | m.9957 T > Cm.9997 T > C | |
| MT-CO1 | m.6597C > A | |
| MT-CYB | m.14787del4 | |
| Mitochondrial Encephalomyopathy | MT-TL1 | m.3242G > A |
| MT-ND6 | m.14487 T > C | |
| MT-ND3 | m.10197G > A | |
| Myoclonic epilepsy with ragged-red fibers (MERRF) | MT-TK | m.8344A > Gm.8356 T > C |
| Lethal Infantile Mitochondrial Disease | MT-TW | m.5567 T > C |
| Leber's Hereditary Optic Neuropathy (LHON) | MT-ND1 | m.3460G > A |
| MT-ND4 | m.11778G > A | |
| MT-ND6 | m.14484 T > C | |
Some of the papers have suggested that alcohol and tobacco consumption are one of the main environmental factors which trigger vision loss in LHON.99–104 Tobacco smoke contains mixture of toxic contents including free radicals which make alterations in the ETC mechanism and it could make a synergic effect with alcohol in the LHON condition.99 Reports showed that persons who have mt.3460 and mt.14484 LHON mutations have high chances to get disease condition by high consumption of alcohol and tobacco102,105,106 reported that smoking is significantly associated with disease penetrance, degree of smoking and number of years smoked correlated with increased risk of developing symptoms. In contradictory to this reports, a study of affected and unaffected siblings from 80 sibships with LHON did not show any significant deleterious association between alcohol or tobacco consumption with vision loss in diseased condition.107
Other than the tobacco and alcohol consumption, some other environmental factors are also involved in the epigenetic mechanism of LHON. Antiretroviral drugs of the nucleoside analog (zidovudine) class are known to be associated with mitochondrial toxicity.108 Clinical studies are reported in some individual cases of LHON associated with antiretroviral therapy for human immunodeficiency virus (HIV) infection.109–111 HIV patients with family history of LHON who require antiretroviral therapy are more triggered for the severe vision loss.112 Je Hyun Seo et al. proposed antituberculosis medication might be an epigenetic factor of LHON in patients with primary mutation.113 In one of the case, report showed clinical expression of LHON precipitated by carbon monoxide poisoning who have mitochondrial primary mutation.114 Long exposure to n-hexane and other organic solvent uncouples mitochondrial respiration and impairs ATP synthesis, this suggest that these compounds could be a triggering epigenetic factor for LHON.115 Occupational exposure to polycyclic aromatic hydrocarbons (PAH) may provide pro-oxidative effect to the mitochondrial dysfunction for the person who has primary LHON mutation and it causes vision loss. Studies have been reported vision loss by LHON after the person had been exposed to rubber tire fire and suggesting that it could be a factor to trigger epigenetic mechanism in LHON.116,117 More validated data and research studies are required to confirm these factors and epigenetics as a cause for LHON. Understanding and identification of clinical and environmental factors and its mechanism of epigenetic changes would help to open a novel therapeutic method and more understanding of the disease condition.
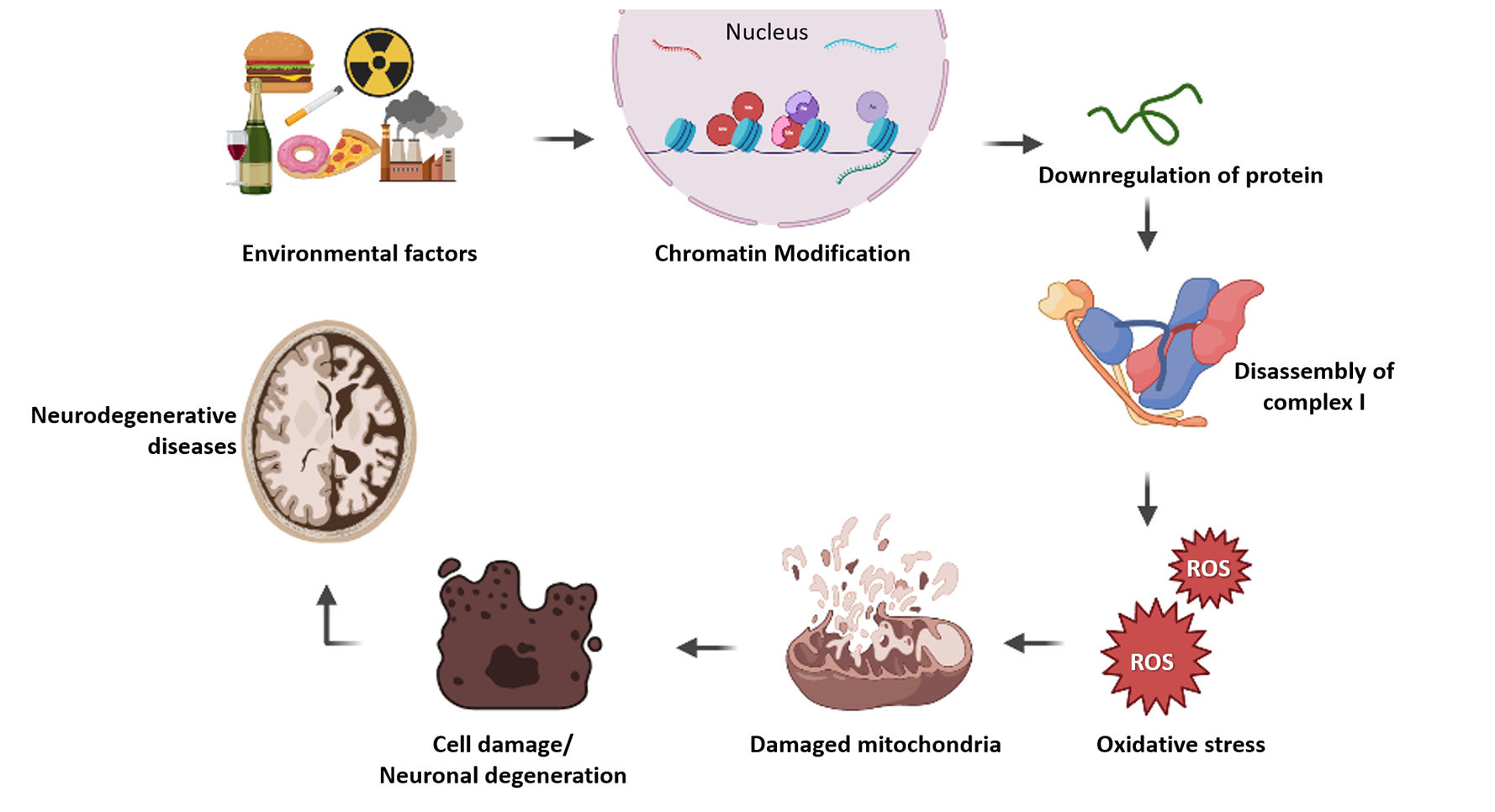
Epigenetic modification of nuclear genes involved in mitochondrial diseasesAccording to proteomics survey, approximately 67% of mitochondrial proteins are present in the mitochondrial matrix, followed by 21% located within the inner mitochondrial membrane, 6% of proteins reside in the inner membrane space and 4% present in the outer mitochondrial membrane and 99% of these mitochondrial proteins are encoded by nuclear genome.118 Changes in mitochondrial protein expression leads to defective oxidative phosphorylation and ATP synthesis which leads to cell death and energy deficiency in cells. This metabolic changes could be the reason for many mitochondrial diseases and other complex disorders. nDNAs are protected with histones but not in mtDNA, that is the major difference between these two genomes. Post-translational histone modification and various DNA methylations regulate nuclear gene expression and protein synthesis in the cell. This kind of epigenetic change will also happen for nuclear encoded mitochondrial genes, which results in decreased level of mitochondrial proteins and leads to dysfunction of mitochondria. Adequate epigenetic studies help to perceive the relevance of each mitochondrial protein associated with the complex diseases. Researches in epigenetic regulation of nuclear encoded mitochondrial genes will lead to better understanding of various diseases and increases the chances for better therapeutics in future. But the epigenetic research is still not much explored in nuclear encoded mitochondrial proteins in disease conditions (Fig. 1).

Illustration of how epigenetics involved in mitochondrial disease progression. Epigenetic factors modify histone proteins in chromatin and it affects gene regulation. Altered gene expression leads to depletion in protein level which results improper assembly of subunits in complex 1. This will affect the functioning of mitochondria and oxidative stress takes place. Mitochondrial damage leads to cell death and it causes various degenerative diseases.
Manganese Superoxide Dismutase (SOD2), encoded by nuclear gene sod2, is an important mitochondrial antioxidant enzyme whose activity has broad implications in health and disease. Many studies have been done in epigenetic regulation of this gene in cancer biology with free radical theory. Studies have reported that reduced SOD activity with CpG methylation in the intronic enhancer region119 and with reduced H3K4me2 histone modification at intronic enhancer which leading to the altered expression of MnSOD.120 In diabetic condition , sod2 is modified with methyl H4K20 and H3K9 acetylation in the promoter region.121 H3K4 histone methylation of retinal sod2 gene has crucial role in development of diabetic retinopathy.122 These studies raise the possibility to use therapeutic targeted towards regulation of methylation status of histones to prevent inhibition of MnSOD and protect mitochondrial damage. In paraganglioma disease condition, succinate dehydrogenase(SDH) gene expression downregulated with H3K27me3 histone modification in the nuclear gene and it can be reversed by overexpression of the JMJD3 histone demethylase.123 Abnormal hypermethylation in the promoter region of the VDAC (Voltage Dependent Anion Channels) gene shows reduced expression of VDAC porin protein and it causes low sperm motility in athenospermia condition in men.124 Neurodegenerative diseases are more with multifactorial etiology having a very high ability to make changes in quality and standards of life.125–128 Along with mitochondrial dysfunctions and epigenetics changes, evidences are indicating that various metabolic pathways have been involved in progression and development of neurodegenerative condition.129–132
Epigenetic regulation of nuclear encoded mitochondrial gene has an important role in the disease progression and cell functioning. Studies have been focused on various aspects of epigenetics play a distinguished role in various ocular neurodegenerative diseases.133 Downregulation of mitochondrial proteins leads to abnormal metabolic condition in cell through damaged oxidative phosphorylation and free radical formation. Cell-based approaches on ocular diseases are a new dimension towards treatment approaches treatment.134 This condition will cause cell death and leads to severe diseases. But still more research is needed to clarify the actual mechanisms. So understanding of epigenetic mechanism and designing therapeutic for the epigenetic targets will improve the treatment efficiency in various diseases.
ConclusionThe purpose of this review was to find out the possible ways of how epigenetics can affect the process of mitochondrial oxidative phosphorylation leading to various diseases focusing on ocular diseases. There is a solid incentive to know more about epigenetics and reconnoiter the role of epigenetics in terms of mitochondrial diseases because these are the factors that has the potential to understand variable penetrance, phenotypic heterogeneity and environmental factors that induce disorders from this group. The current technology has the ability to directly evaluate epigenetic discrepancy in the nuclear genome or mitochondria. These situations have ultimately led to the urge of being able to find a permanent solution. Therefore, our focus is to develop on the idea of epigenetic modifications involved in mitochondrial diseases leading to various optical neuropathies, especially LHON.
FundingThe authors would like to thank the Science and Engineering Research Board (SERB) for providing Early Career Research Award (ECR/2018/000718), India to complete this article successfully.
