



Las ataxias (AT) y paraparesias espásticas hereditarias (PEH) son síndromes neurodegenerativos raros. Nos proponemos conocer la prevalencia de las AT y PEH en España en 2019.
Pacientes y métodosEstudio transversal, multicéntrico, descriptivo y retrospectivo de los pacientes con AT y PEH, desde marzo de 2018 a diciembre de 2019 en toda España.
ResultadosSe obtuvo información de 1933 pacientes procedentes de 11 Comunidades Autónomas, de 47 neurólogos o genetistas. Edad media: 53,64 años±20,51 desviación estándar (DE); 938 varones (48,5%), 995 mujeres (51,5%). En 920 pacientes (47,6%) no se conoce el defecto genético. Por patologías, 1.371 pacientes (70,9%) diagnosticados de AT, 562 diagnosticados de PEH (29,1%). La prevalencia estimada de AT es 5,48/100.000 habitantes, y la de PEH es 2,24 casos/100.000 habitantes. La AT dominante más frecuente es la SCA3. La AT recesiva más frecuente es la ataxia de Friedreich (FRDA). La PEH dominante más frecuente es la SPG4, y la PEH recesiva más frecuente es la SPG7.
ConclusionesLa prevalencia estimada de AT y PEH en nuestra serie es de 7,73 casos/100.000 habitantes. Estas frecuencias son similares a las del resto del mundo. En el 47,6% no se ha conseguido un diagnóstico genético. A pesar de las limitaciones, este estudio puede contribuir a estimar los recursos, visibilizar estas enfermedades, detectar las mutaciones más frecuentes para hacer los screenings por comunidades, y favorecer los ensayos clínicos.
Ataxia and hereditary spastic paraplegia are rare neurodegenerative syndromes. We aimed to determine the prevalence of these disorders in Spain in 2019.
Patients and methodsWe conducted a cross-sectional, multicentre, retrospective, descriptive study of patients with ataxia and hereditary spastic paraplegia in Spain between March 2018 and December 2019.
ResultsWe gathered data from a total of 1933 patients from 11 autonomous communities, provided by 47 neurologists or geneticists. Mean (SD) age in our sample was 53.64 (20.51) years; 938 patients were men (48.5%) and 995 were women (51.5%). The genetic defect was unidentified in 920 patients (47.6%). A total of 1371 patients (70.9%) had ataxia and 562 (29.1%) had hereditary spastic paraplegia. Prevalence rates for ataxia and hereditary spastic paraplegia were estimated at 5.48 and 2.24 cases per 100 000 population, respectively. The most frequent type of dominant ataxia in our sample was SCA3, and the most frequent recessive ataxia was Friedreich ataxia. The most frequent type of dominant hereditary spastic paraplegia in our sample was SPG4, and the most frequent recessive type was SPG7.
ConclusionsIn our sample, the estimated prevalence of ataxia and hereditary spastic paraplegia was 7.73 cases per 100 000 population. This rate is similar to those reported for other countries. Genetic diagnosis was not available in 47.6% of cases. Despite these limitations, our study provides useful data for estimating the necessary healthcare resources for these patients, raising awareness of these diseases, determining the most frequent causal mutations for local screening programmes, and promoting the development of clinical trials.
Las ataxias (AT) y paraparesias espásticas hereditarias (PEH) son síndromes neurodegenerativos de base genética altamente heterogénea. La prevalencia es desconocida, pero se estima en unos 3-20 enfermos por cada 100.000 habitantes para las ataxias, y 1,2-9,6 por cada 100.000 habitantes en el caso de las paraparesias espásticas, encuadrándose dentro de las enfermedades raras1–3.
Actualmente en España no se conoce la prevalencia real de estas dos enfermedades. Se han publicado series de pacientes por comunidades autónomas o regiones en las últimas dos décadas, con pequeñas muestras aisladas de pacientes4–8. A nivel internacional hay algunas publicaciones multicéntricas que intentan, con sus limitaciones, dar una aproximación de la prevalencia de estas patologías9–14. Estos estudios muestran prevalencias distintas, influidas por una metodología variable, una clasificación también heterogénea de los pacientes, y diferentes criterios de inclusión. Actualmente no existen screenings de muestras poblacionales, los estudios de prevalencia suelen salir de regiones con agrupaciones de casos, y los estudios publicados son mayoritariamente de países europeos, no habiendo estudios de prevalencia en países norteamericanos, africanos, o australianos.
Un correcto estudio epidemiológico requiere de una recogida de datos exhaustiva, «puerta a puerta», de cada paciente, para obtener el mayor número de casos y un tamaño muestral óptimo, con consentimientos expresos de todos los pacientes para la recolección de datos y estudio posterior de sus muestras, y conlleva el análisis de todas las variables clínicas y genéticas relacionadas, la evolución temporal, y la relación familiar entre el caso índice y los descendientes.
En nuestro caso, no se ha podido emplear esta metodología por la complejidad de la misma, por lo que hemos asumido las limitaciones del diseño de un mapa transversal retrospectivo, y hemos realizado un estudio de aproximación a la prevalencia de estas enfermedades en España.
No existe en este momento un mapa epidemiológico de los tipos de AT y PEH (APEH) en nuestro país. Esta información es de vital importancia para el desarrollo de políticas sanitarias, ubicar y dotar unidades de referencia con criterios adaptados a las necesidades, favorecer la atención en centros especializados, identificar estrategias de investigación, y planificar ensayos clínicos. Una gestión más eficiente de los recursos beneficiará a todo el sistema, sin perder el enfoque en la mejor atención al paciente15. Nuestro objetivo con este estudio consiste en conocer de forma aproximada la prevalencia de APEH en España en 2019 y la distribución geográfica de las mutaciones y genes implicados.
Material y métodosEstudio epidemiológico multicéntrico, descriptivo, retrospectivo y transversal de la prevalencia de las APEH en España en el año 2019 (mapa diagnóstico). El objetivo es conocer la prevalencia de las diferentes formas de APEH, y su distribución en las diferentes comunidades autónomas. Los datos procedentes de pacientes sin diagnóstico genético se han analizado separadamente.
Se ha contactado con las Unidades de Referencia (CSUR) de Ataxias y Paraplejias Hereditarias en España, y con la mayoría de las Unidades de Ataxias de los principales hospitales de cada comunidad autónoma. Así mismo, a través de la Sociedad Española de Neurología, mediante correos electrónicos y avisos en la página web, se ha invitado a participar a neurólogos, neuropediatras, o genetistas miembros de la SEN con ejercicio profesional en territorio español, que atendieran pacientes con APEH en sus consultas, y estuvieran dispuestos a participar.
Se han incluido pacientes mayores de 16 años y menores de 60 años, siendo el periodo de reclutamiento desde marzo de 2018 hasta diciembre de 2019, ambos inclusive. Se han excluido los casos adquiridos.
Las variables registradas han sido: edad, sexo, lugar y fecha de nacimiento, fecha de inicio de síntomas, centro de procedencia, fenotipo, antecedentes familiares, estudio genético y mutación (en los casos posibles). Hemos valorado también los casos con alta sospecha de origen hereditario con o sin historia familiar, y con estudio genético negativo, o con resultados no concluyentes o variantes de significado incierto.
Las limitaciones del diseño son las inherentes a todo estudio epidemiológico descriptivo, transversal, retrospectivo, además de las inherentes al estudio de patologías minoritarias. Al ser datos anonimizados, el riesgo de duplicidad o solapamiento de individuos es uno de los principales sesgos de selección que podemos encontrar. Para intentar corregirlo, hemos incluido la fecha de nacimiento de cada paciente como factor para controlar las duplicidades. Las limitaciones de la fuente de información son los registros y bases de datos sobre estas enfermedades disponibles en los centros y comunidades autónomas. En muchos casos o no existen, o son muy rudimentarios. Las técnicas diagnósticas de los laboratorios actuales no son comparables a las disponibles hace 10 o 20 años, lo que puede influir en el número de casos con genética indeterminada.
Los datos se han tratado de forma anonimizada y agrupada, y se han almacenado en una base de Microsoft Excel, después formalizada en un documento del programa SPSS en su versión 25 de IBM Statistics, a través del cual se ha realizado el análisis estadístico. Para los cálculos de prevalencia estimada, hemos tomado en consideración que en España, a 1 de enero de 2019, se encontraban censados 47.007.367 habitantes, según datos del Instituto Nacional de Estadística16. Del total, 23.033.803 eran hombres, y 23.973.564 eran mujeres. Para nuestros cálculos, hemos considerado las cifras de población comprendida entre los 16 y los 60 años (25.017.427 millones de habitantes).
Este proyecto ha sido evaluado favorablemente por el Comité de Ética y Deontología de la Sociedad Española de Neurología (febrero de 2018), así como por los distintos Comités de Ética de la mayoría de las Comunidades Autónomas participantes.
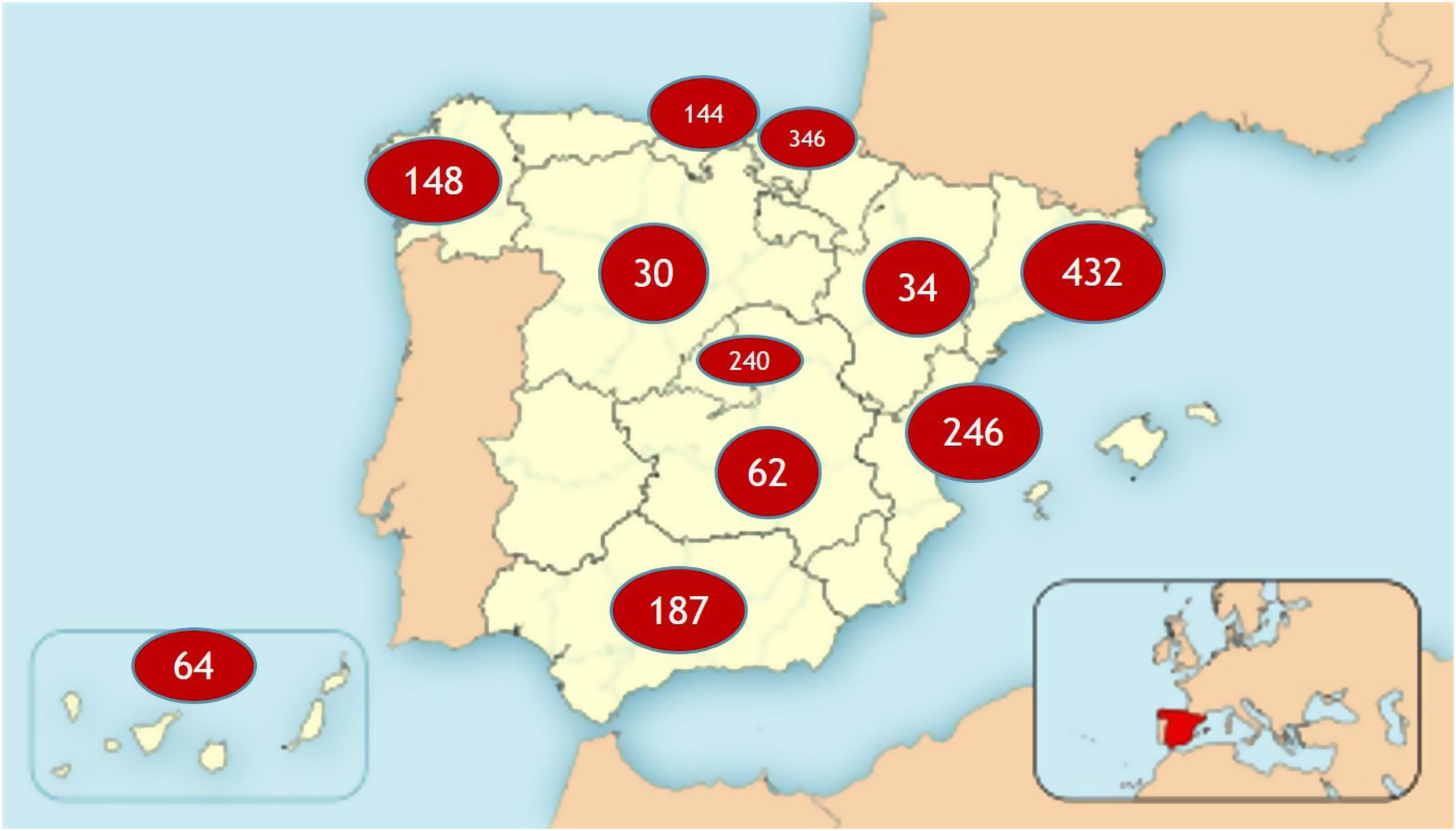
ResultadosRegistramos un total de 1.933 pacientes de 11 Comunidades Autónomas (CC. AA.), con participación de hospitales de diferente tamaño, CSUR de Ataxias y Paraplejias Hereditarias nacionales, y un total de 47 neurólogos o genetistas (fig. 1). La prevalencia estimada total de APEH sería de 7,73 por 100.000 habitantes.

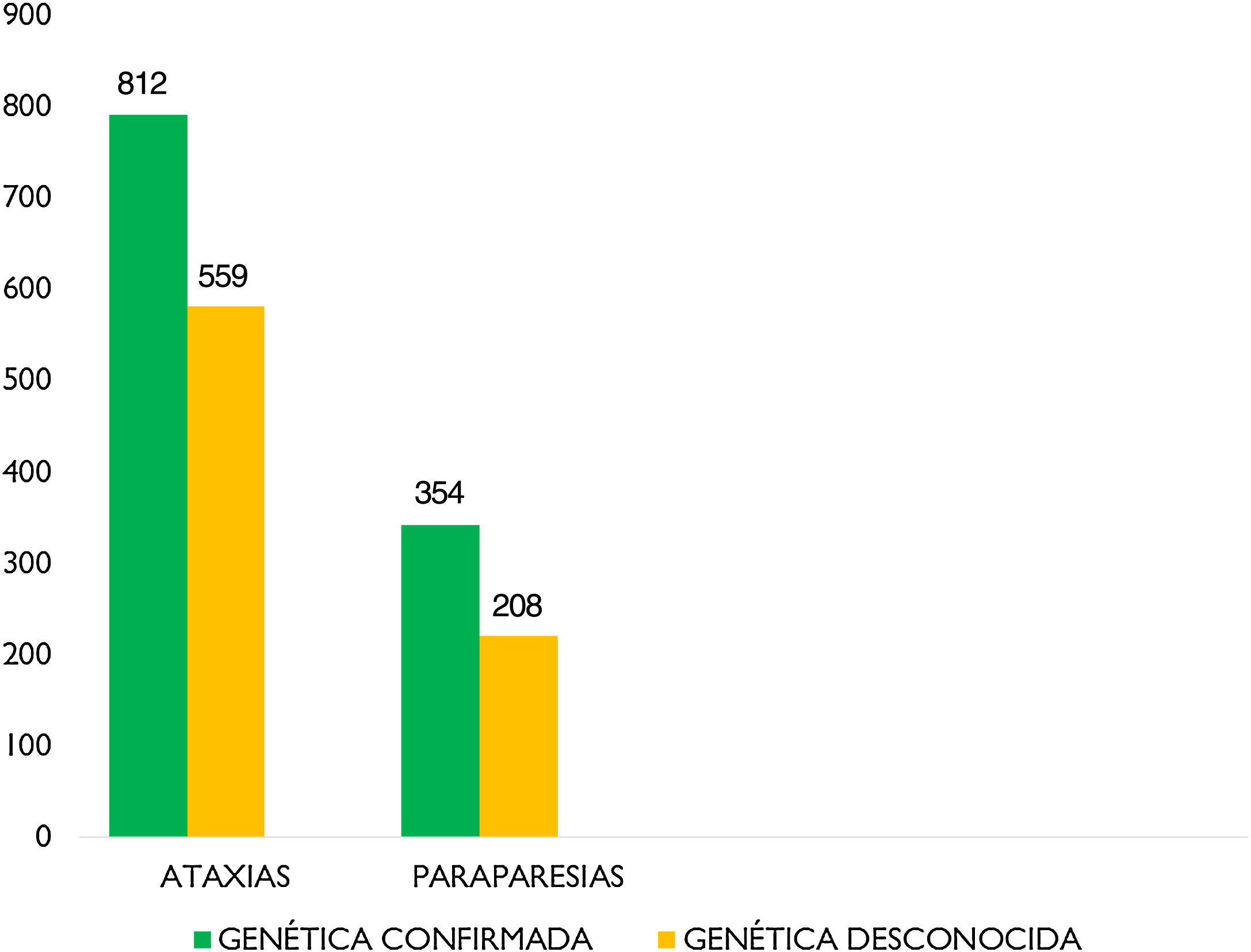
La edad media es de 53,64 años±20,51 DE, con una mediana de 54 años. Del total, son varones 920 pacientes (50,8%) y 889 son mujeres (49,2%). En 920 pacientes (47,6%) no se conoce el defecto genético subyacente (fig. 2).

Por patologías, 1.371 pacientes (70,9%) han sido diagnosticados de AT, y 562 (29,1%) han sido diagnosticados de PEH (29,1%) (tabla 1).
Subtipos de AT y PEH en nuestra serie (entre paréntesis los casos índice de cada grupo)
| N | AT dominante | AT recesiva | Otras ataxias | PEH dominante | PEH recesiva | Otras PEH | |
|---|---|---|---|---|---|---|---|
| SCA3 131 casos (47) | FRDA 249 casos (232) | FXTAS 20 casos (19) | SPG4 150 casos (80) | SPG7 56 casos (41) | ABCD1 11 casos (11) | ||
| SCA 2 47 casos (36) | NPC1 18 casos (16) | Pelizaeus-Merzbacher 1 caso (1) | SPG17 32 casos (10) | SPG11 22 casos (21) | 5PG2 1 caso (1) | ||
| SCA6 37 casos (27) | AOA2 21 casos (13) | Kearns-sayre 1 caso (1) | SPG10 21 casos (13) | SPG5A 5 casos (5) | |||
| SCA1 37 casos (29) | AT-T 17 casos (12) | SPG3A 14 casos (12) | SPG15 4 casos (4) | ||||
| SCA37 34 casos (14) | SPAX6 12 casos (9) | SPG31 11 casos (8) | SPG35 4 casos (4) | ||||
| AE2 35 casos (25) | Xantomatosis cerebrotendinosa 12 casos (8) | SPG13 3 casos (2) | SPG48 4 casos (2) | ||||
| SCA36 24 casos (15) | SCAR8/ARCA 1 9 casos (7) | ODD 2 casos (2) | SPG76 4 casos (3) | ||||
| SCA7 15 casos (14) | POLR3A 6 casos (5) | SPG 12 1 caso (1) | SPG54 3 casos (1) | ||||
| SCA48 7 casos (1) | SCAR10 6 casos (3) | SPG20 2 casos (1) | |||||
| SCA17 6 casos (6) | Déficit Vit E 5 casos (5) | SPG30 2 casos (1) | |||||
| SCA19 6 casos (3) | POLG1 4 casos (4) | SPG43 1 caso (1) | |||||
| AE1 6 casos (6) | AOA4 3 casos (3) | SPG46 1 caso (1) | |||||
| SCA8 5 casos (5) | SPAX4 2 casos (1) | ||||||
| SCA23 2 casos (2) | Enf. Sust. Blanca evanescente 2 casos (1) | ||||||
| SCA35 2 casos (2) | Sd.Cockayne A 2 casos (1) | ||||||
| SCA40 2 casos (1) | Sd. Cockayne B 2 casos (1) | ||||||
| DRPLA 2 casos (2) | SPAX 2 1 caso (1) | ||||||
| Alexander 2 casos (2) | SPAX5 1 caso (1) | ||||||
| SCA13 1 caso (1) | PCARP 1 caso (1) | ||||||
| SCA12 1 caso (1) | SCAR 16 1 caso (1) | ||||||
| SCA10 1 caso (1) | Déficit metilación homocisteína 1 caso (1) | ||||||
| OPA1 1 caso (1) | Wilson 1 caso (1) | ||||||
| AE5 1 caso (1) | Sandhoff 1 caso (1) | ||||||
| A.cerebelosa+ migraña hemipléjica 1 caso (1) | POLR3B 1 caso (1) | ||||||
| PMM2 1 caso (1) | |||||||
| Brown-Vialetto 1 caso (1) | |||||||
| Marinesco-Sjogren 1 caso (1) | |||||||
| TOTAL | 406 (294) | 384 (336) | 22 (21) | 234 (171) | 108 (85) | 12 (12) |
AT-T: ataxia-telangiectasia.
La prevalencia estimada de AT en nuestra serie, con los datos de las comunidades participantes, se encuentra en 5,48 casos por 100.000 habitantes (en AT dominantes 1,62/100.000 habitantes, y en AT recesivas la prevalencia estimada es de 1,53/100.000 habitantes).
La prevalencia estimada de PEH en la misma franja de edad es de 2,24 casos por 100.000 habitantes (las PEH dominantes tienen una prevalencia estimada de 0,93/100.000 habitantes, y las PEH recesivas tienen una prevalencia estimada de 0,43/100.000 habitantes) (tabla 2).
Prevalencia estimada según series publicadas
| APEH | PEH | AH | Otras | |
|---|---|---|---|---|
| Ruano, 2014 (mundial) | 9,8/105 | 3,6/105 | 6/105 | - |
| **Polo, 1991 (Cantabria) | 20,2/105 (índ: 9,4/105) | 9,6/105 | 10,6/105 | - |
| **Infante, 2005 (Cantabria) | - | - | - | AH-AD: 1,6/105 |
| Erichsen, 2009 (Noruega) | 13,9/105 | 7,4/105 | 6,5/105 | - |
| Braschinsky, 2009 (Estonia) | - | 4,4/105 | - | - |
| Orsucci, 2014 (Toscana) | - | 2,2-3,4/105 | - | - |
| Hutchinson, 2002 (Irlanda) | - | - | - | PEH-AD «pura»: 1,27/105 |
| Zortea, 2004 (Padua) | - | - | 9,3/105 | - |
| Coutinho, 2013 (Portugal) | 13/105 | 4,1/105 | 8,9/105 | - |
| Mapa APEH CEAPED 2019 (España) | 7,73/105 | 5,48/105 | 2,24/105 |
Por subtipos, y valorando solo los casos con genética confirmada: dentro de los 406 casos de AT dominantes, las más frecuentes son la SCA3 (131 casos), y la SCA2 (47 casos). De las 384 pacientes con AT recesivas, las más frecuentes son la FRDA (249 casos), y NPC1 (18 casos). En cuanto a las PEH, de las 234 dominantes, las más frecuentes son SPG4 (150 pacientes) y SPG17 (32 casos); de los 108 casos de PEH recesivas las más frecuentes son SPG7 (56 casos), y SPG11 (22 casos) (tabla 1).
DiscusiónConocer la prevalencia de una enfermedad rara es un reto para cualquier sistema sanitario o grupo poblacional17. Su baja prevalencia, su dispersión geográfica y su elevada complejidad diagnóstica, unido a la dificultad de acceso a los test necesarios, especialmente los genéticos, las convierten en enfermedades poco visibles de difícil cuantificación18–27.
En este estudio hemos intentado salvar algunas de esas dificultades para conocer la prevalencia de las APEH en España.
La iniciativa nace en el año 2017 de la mano de la Comisión de Estudio de Ataxias y Paraparesias Espásticas (CEAPED) de la Sociedad Española de Neurología (SEN) que proyectó la realización de un mapa transversal, para poder estimar una prevalencia de las APEH en España. Durante los siguientes dos años se ha realizado un esfuerzo organizativo desde la SEN y la CEAPED para poder contactar y transmitir este proyecto al mayor número de neurólogos posible, además de genetistas y neuropediatras. Es un trabajo de screening con limitaciones claras, por el tipo de diseño del estudio, la dificultad de organización de un trabajo multicéntrico, y la exclusión de casos menores de 16 años. Sin embargo, creemos que la generosa colaboración de los neurólogos participantes, el acceso a todos ellos mediante correo desde la SEN, la simplicidad del registro y el trabajo conjunto coordinado, han hecho que los datos obtenidos sean numerosos y valiosos y pueden ser comparados con hallazgos previos en nuestro entorno y en series procedentes de otros países.
Así, en nuestro país, el primer intento de registro multicéntrico para estas entidades fue el Registro Español de Ataxias y Paraparesias espásticas degenerativas (REDAPED), puesto en marcha en 2012 por un grupo de expertos nacionales, apoyado por el Instituto de Investigación de Enfermedades Raras del Instituto Carlos III, con unos objetivos rigurosos y ambiciosos. Actualmente este proyecto se encuentra parado por distintos factores, por lo que lamentablemente no podemos desglosar sus resultados ni son accesibles para comparación con los actuales.
Disponemos de cuatro series nacionales publicadas hasta la fecha, tres de ellas del grupo de Cantabria: en 1991 publicaron 48 casos índice de AT y PEH registrados solo en sus CC. AA. en una década4–8. Principalmente tenían 18 familias de FRDA, y 9 familias con PEH. Describían entonces una prevalencia de APEH de 20,2 casos por 100.000 habitantes. En la segunda, del año 1999, describen una serie de 87 familias, y 60 casos esporádicos, pero solo son casos de AT (pertenecientes a 9 CC. AA.), donde la SCA2 y SCA3 tenían una prevalencia del 15%, y el 58% de sus casos eran genéticamente inclasificables. La tercera serie está centrada en las AT autosómicas dominantes en Cantabria, y obtienen 30 familias, principalmente SCA2 y SCA3, y no encuentran mutación concreta en 9 familias (30%). Su prevalencia estimada es de 1,6 casos por 100.000 habitantes. La cuarta serie nacional, del año 2000, describe 121 pacientes solo con AT dominantes y FRDA, de 5 a 80 años de edad, de 8 CC. AA. La SCA3 fue la AT dominante más frecuente, seguida de SCA7, y mucho menos frecuente la SCA2. El 41,2% de las AT dominantes, y el 43,5% de las AT recesivas no tenían genética determinada.
Al intentar comparar nuestros resultados con estas series, nos encontramos con dos limitaciones importantes: solo uno de los artículos incluye AT y PEH, y al haber pasado más de 20 años de su publicación, los estudios genéticos y métodos de diagnóstico y registro han cambiado de forma significativa, por lo que son difícilmente comparables.
Respecto a datos generales, en 2014 se publicó una revisión sistemática que incluyó 22 estudios publicados entre 1983 y 2013, con un total 14.539 pacientes de 16 países distintos13. En esta extensa revisión, se reflejó una prevalencia estimada de la AT dominante de 5,6/100.000 habitantes, y de la recesiva de 3,3/100.000 habitantes (no todos los casos con genética confirmatoria). La prevalencia estimada en nuestra serie fue de 1,62/100.000 habitantes en AT dominantes, y de 1,53/100.000 habitantes en AT recesivas. En cuanto a la prevalencia de las PEH, en esa revisión se describió una prevalencia de PEH autosómicas dominantes de 5,5/100.000 habitantes, y de PEH autosómicas recesivas de 5,3/100.000 habitantes, frente a las prevalencias de 0,93/100.000 habitantes, y 0,43/100.000 habitantes respectivamente de nuestra serie.
A pesar del importante número de casos recogidos, la disminución de la prevalencia de nuestra serie frente a las publicadas, tanto en AT como en PEH, puede deberse a varios factores: la limitación en el rango de edades que pusimos para la recepción de datos (de 16 a 59 años, excluyendo los casos infantiles, y a los mayores de 60 años, por la posible coexistencia de otras causas de ataxia como patología cerebrovascular, tumoral, etc.). Aunque hemos conseguido una excelente colaboración y comunicación con la mayoría de las CC. AA., algunas no han podido participar, lo que ha limitado el número total de casos. También los factores locales, como la limitación del acceso a test genéticos, el diferente acceso de los pacientes a los medios y unidades o especialistas en sus respectivas CC. AA. ha podido ser otro de los factores limitantes.
Si analizamos las prevalencias de los diferentes subgrupos en nuestra serie, la SCA3 fue la ataxia autosómica dominante más frecuente mientras que la ataxia de Friedreich fue la forma recesiva más prevalente, de forma similar a lo hallado en publicaciones previas. Sin embargo, destaca el elevado número de pacientes con SCA36, SCA37 y NPC1 identificados en este estudio. Estas diferencias se explican por el hecho de que SCA36 y SCA37 han sido originariamente descritas en España lo que ha llevado a la identificación de un mayor número de casos en el contexto de las investigaciones iniciales y a una posterior sensibilización para ese diagnóstico en las áreas geográficas españolas donde se describieron. Respecto a NPC1 creemos que las cifras guardan relación con el interés específico de algunos grupos locales en esta enfermedad, lo que ha llevado a aglutinar a un mayor número de casos en dichos centros que, por su parte, han colaborado en el registro activamente.
Respecto a las paraparesias, la revisión del 2014 mencionada muestra que la forma dominante más frecuente fue SPG4, seguida de SPG3, y entre las PEH recesivas, la más frecuente fue SPG11, seguida de SPG15. Estos datos son similares a otras publicaciones internacionales de menor tamaño muestral8,9. En nuestra serie, SPG4 fue la forma dominante más frecuente, y en las recesivas SPG7 fue la más prevalente, acorde con dichas publicaciones. Por su parte SPG15 ocupó la cuarta posición en nuestra serie con solo 4 casos índices identificados.
Finalmente el estudio ha mostrado que un 47,6% de nuestros pacientes con APEH no disponen de un diagnóstico genético definitivo. Esta cifra está dentro del rango de lo publicado para estas entidades (33% a 92%)18–28, y se podría mejorar con una mejor implementación de los estudios genéticos.
ConclusionesLa prevalencia estimada de AT en nuestra serie es de 5,48 casos/100.000 habitantes, y la de PEH es de 2,24 casos/100.000 habitantes. La AT dominante más frecuente es la SCA3, seguida de la SCA2. Las AT recesivas más frecuentes son la ataxia de Friedreich, y el Niemann-Pick C. La PEH dominante más frecuente es la SPG4, seguida de la SPG17, y la PEH recesiva más frecuente es la SPG7, seguida de la SPG11.
Estas frecuencias son similares a las del resto del mundo. En el 47,6% no se ha conseguido un diagnóstico genético. A pesar de las limitaciones, esta es la mejor información disponible sobre la prevalencia, frecuencia y distribución geográfica de estas enfermedades en nuestro país.
El diagnóstico genético es imprescindible para mejorar la asistencia médica cotidiana, visibilizar estas enfermedades, detectar las mutaciones más frecuentes para hacer los screenings más adecuados por zonas o comunidades, optimizar los registros estatales, evaluar la necesidad de recursos, diseñar ensayos clínicos y facilitar el reclutamiento de pacientes para los mismos.
FinanciaciónLos resultados parciales de este estudio han sido presentados en las Reuniones Anuales de la Sociedad Española de Neurología en 2017 y 2018, y financiado parcialmente en forma de beca por la Sociedad Española de Neurología a la autora principal (Dra. Gloria Ortega Suero), encargada de la realización de la base, y la custodia de datos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

