




Multiple endocrine neoplasia type 1 (MEN 1) is a rare, autosomal dominant hereditary disease with a prevalence of approximately 2 cases per 100,000 inhabitants, attributable to a mutation of the MEN1 gene located on chromosome 11q13, and which encodes for menin (a nuclear cell cycle regulating protein). This mutation predisposes to the development of tumors, particularly of the parathyroid glands, enteropancreatic endocrine cells and anterior pituitary, but also in numerous other locations, including the adrenal cortex. The penetrance is so high (about 98% of all affected patients will develop some neoplasm in the first 5 decades of life) that strict follow-up is recommended from the age of 5 years. The same mutation may result in different phenotypes in each affected relative. The development of hyperandrogenism in a patient with MEN 1 at any age requires the exclusion of an adrenal gland tumor.1
We report the case of a boy subjected to follow-up from 5 years of age due to the identification of carrier status of the p.ala337asp mutation of the menin gene as the result of a family study (father with the same mutation, subjected to surgery due to a parathyroid adenoma at 30 years of age). At 7 years of age the patient presented incipient pubarche, a thickening of the penis (30×15mm) with no increase in testicle size (2mL) and hypergrowth (weight 26.3kg, height 131.7cm [p92]), a body mass index [BMI] of 15.16kg/m2 (p28), and a growth rate of 8.6cm/year (p>99). The bone age was advanced by 1.5 years. An abdominopelvic MRI scan was reported as normal, and a hormone study was made, with the following results: 17-hydroxyprogesterone 55.7nmol/L (normal<5), total testosterone 0.6nmol/L (normal<0.7), dehydroepiandrosterone sulphate 4.8μmol/L (normal<4.0), androstenedione 8.9nmol/L (normal<4.2), LH 0.1U/L and FSH 0.3U/L. The biochemical profile was consistent with congenital adrenal hyperplasia. Treatment was started with hydrocortisone 15mg/m2/day in 3 doses, followed by a drop in growth rate to 4.8cm/year (p15), as well as a decrease in androgens (testosterone 0.3nmol/L, dehydroepiandrosterone sulphate 3.6mmol/L, androstenedione 4.3nmol/L) and 17-hydroxyprogesterone (12nmol/L). The study of the 21-hydroxylase gene (CYP21A2 gene, located on chromosome 6p21) identified two compound heterozygosis mutations (Ile173Asn/Val282Leu) in the patient and one in each of the parents: Ile173Asn in the father and Val282Leu in the mother. With the late diagnostic confirmation of congenital adrenal hyperplasia due to 21-hydroxylase deficiency, hydrocortisone treatment has been continued to prevent bone age progression, together with the application of the MEN1 neoplasm screening protocol.
Between 20 and 70% of all patients with MEN 1 develop some adrenal gland lesion during their lifetime, and exceptionally in childhood. Most of these lesions are benign, unilateral and non-functioning tumors, but some tumors secreting cortisol, aldosterone and, more rarely, androgens have been reported, as well as pheochromocytoma. Functioning tumors require treatment, as well as those posing a risk of malignancy: lesions measuring over 4cm in size, and those that grow or exhibit atypical or indeterminate features. A conservative approach is usually adopted in the case of smaller non-functioning tumors, since they usually do not change in appearance or increase in size in the course of follow-up.2–7
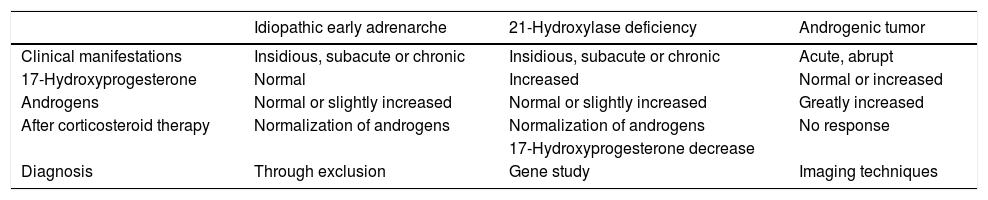
In the event of a child with MEN 1 and hyperandrogenism, the differential diagnosis of the tumor disease should be established with age-inherent conditions such as late congenital adrenal hyperplasia and idiopathic early adrenarche. The hormone profile, the response to corticosteroid treatment, radiological findings and genetic tests establish the diagnosis (Table 1). When a tumor origin is discarded, the existence of some other concomitant disease should be considered. A review of the literature has revealed no previous descriptions of such an association of diseases. Although both are of genetic origin, they are caused by different and distant genes; their association therefore must be entirely casual.
Differential diagnosis of hyperandrogenism in childhood.
| Idiopathic early adrenarche | 21-Hydroxylase deficiency | Androgenic tumor | |
|---|---|---|---|
| Clinical manifestations | Insidious, subacute or chronic | Insidious, subacute or chronic | Acute, abrupt |
| 17-Hydroxyprogesterone | Normal | Increased | Normal or increased |
| Androgens | Normal or slightly increased | Normal or slightly increased | Greatly increased |
| After corticosteroid therapy | Normalization of androgens | Normalization of androgens | No response |
| 17-Hydroxyprogesterone decrease | |||
| Diagnosis | Through exclusion | Gene study | Imaging techniques |
Please cite this article as: García García E, Mangas Cruz MÁ, Guerrero Vázquez R, Navarro González E. Hiperandrogenismo en un niño con neoplasia endocrina múltiple tipo 1. Endocrinol Diabetes Nutr. 2019;66:132–133.
