


Economic losses with high mortality rate associated with Porcine circovirus type 2 (PCV2) is reported worldwide. PCV2 commercial vaccine was introduced in 2006 in U.S. and in 2008 in Brazil. Although PCV2 vaccines have been widely used, cases of PCV2 systemic disease have been reported in the last years. Eleven nursery or fattening pigs suffering from PCV2 systemic disease were selected from eight PCV2-vaccinated farms with historical records of PCV2 systemic disease in Southern Brazil. PCV2 genomes were amplified and sequenced from lymph node samples of selected pigs. The comparison among the ORF2 amino acid sequences of PCV2 isolates revealed three amino acid substitutions in the positions F57I, N178S and A190T, respectively. Using molecular modeling, a structural model for the capsid protein of PCV2 was built. Afterwards, the mutated residues positions were identified in the model. The structural analysis of the mutated residues showed that the external residue 190 is close to an important predicted region for antibodies recognition. Therefore, changes in the viral protein conformation might lead to an inefficient antibody binding and this could be a relevant mechanism underlying the recent vaccine failures observed in swine farms in Brazil.
Porcine circovirus type 2 (PCV2) is a small non-enveloped, icosahedral virus with single-stranded circular DNA that belongs to the Circoviridae family and genus Circovirus. PCV2 has been associated with different disease syndromes collectively named PCV diseases (PCVD) and is considered as one of the most economically important viral pathogens for swine worldwide.1
A genetic diversity within PCV2 strains has been described, although some isolates share high level of nucleotide identity among their genomes.2,3 To date, three different PCV2 genotypes have been determinated4: PCV2a, PCV2b, and PCV2c. In 2012, a variant PCV2 strain, named mutant PCV2b, showing an elongation of ORF2 by one amino acid (Lysine (K)), was detected in several PCVD cases in the United States.5 This variant has been nowadays classified as PCV2d.
An epidemiological transition from sporadic to epidemic PCV2 systemic disease (PCV2-SD) under field conditions, has been suggested, based on a genotype shift of PCV2 isolates over time. PCV2a was predominant on pig farms with sporadic PCV2-SD in several countries prior 2000–2002, while PCV2b has been significantly more prevalent in epidemic PCV2-SD outbreaks from 2002 onwards.4
Besides several indirect control measures, prevention of PCV2-SD has been successfully achieved by means of PCV2 vaccination, since 2006. Up to this moment, several commercial vaccines are currently available; all of them are based on PCV2a strains.6 Nevertheless, it has been shown that genotypes a and b share epitopes and the vaccines induce cross-protective immunity.7,8 In Brazil, the PCV2 vaccination was introduced in commercial pig farms in 2008. However, despite of the continuous use of PCV2 vaccines, some authors have reported PCVD outbreaks in PCV2-vaccinated pigs.9,10 The absence of a consistent model for PCV2-SD reproduction under experimental conditions11 limits the studies to investigate the so called vaccine failures. In this regard, molecular modeling has been widely used to infer important structural information about viral proteins in the absence of experimental data. Over the last years, antigenic studies have identified six linear epitopes (A, B, C, D, E and F) from PCV2 as important sites to antibody recognition.12,13 Also, several studies described a number of amino acids in different regions with possible role in antibody recognition.12,14,15 In this sense, the PCV2 capsid and these epitopes have been used as a target for in silico structural analyses16,17 and in experimental structural studies by Cryo electron microscopy18 and X-ray crystallography.13
Aiming to study the possible alteration in the epitope conformation of the capsid protein of PCV2 owing to mutation, molecular modeling methodology was employed to build a model for the viral capsid from PCV2 isolates implicated in cases of PCV2-SD in vaccinated pig herds in Brazil.
Materials and methodsCase selectionConventional nursery and fattening farms located in Southern Brazil with historical records of PCV2-SD were selected for this study. Pigs were vaccinated against PCV2 once or twice at weaning (21 days-old) with one out of four PCV2-vaccines available in Brazil, according to the farms’ choice. Up to three pigs per farm, totalizing eleven pigs suffering from clinical signs of PCV2-SD, were euthanized and subsequent necropsied.
All selected pigs fulfilled the PCV2-SD diagnosis, according to individual disease case definition criteria that includes1 clinical signs (growth retardation and wasting); moderate to severe histopathological lesions in lymphoid tissues (lymphocyte depletion with granulomatous inflammation); and moderate to high amount of PCV2 within microscopic lesion detected by immunohistochemistry (IHC).19
DNA sequencing and phylogenetic analysisSequencing was performed directly from PCV2 DNA amplified by PCR from lymph node samples from the eleven PCV2-SD affected pigs by the Sanger method using primers described by Mankertz et al.2 and Dupont et al.20 PCR products were gel purified using the MinElute Gel Extraction kit (Qiagen, Hilden, Germany), amplified using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and then purified with BigDye XTerminator Purification Kit (Applied Biosystems, Foster City, CA, USA). The nucleotide sequences were determined using an ABI3130xl Genetic Analyzer.
The obtained sequences were analyzed and assembled with the Phred/Phrap/Consed softwares.21,22 For the phylogenetic analysis, 46 PCV2 complete genome sequences, including other representative sequences of PCV2a, PCV2b, PCV2c and PCV2d available in GenBank database (http://www.ncbi.nlm.nih.gov/GenBank) were aligned using ClustalW in MEGA 6.0 software.23 Phylogenetic relationships among sequences were analyzed in MEGA 6.023 using the Maximum Composite Likelihood method. Confidence in the Neighbor-Joining (NJ) tree was estimated by 1000 bootstrap replicates.
Furthermore, amino acid sequence of the capsid protein of the eleven PCV2 isolates (Genbank accession number KT719404, KT819159, KT819160, KT819162, KT819163, KT819164, KT819165, KT819166, KT819167, KT819169 and KT819170) were aligned to the PCV2 crystallographic structure (Protein Data Bank ID: 3R0R:A) using the Clustal program24 and plotted by EPSprint server.25
Molecular modeling of PCV2 capsid proteinA PCV2 sequence (KT719404) was selected to generate a representative model of the capsid protein monomer of the PCV2 by I-TASSER online server.26 This model was built using as template the PCV2 monomer consensus sequence (PCV2CS) structure (PDB ID: 3R0R:A) without the N-terminal portion.13 Although the PDB structure does not contain the N-terminal portion in comparison with cryo-electron microscopy (Cryo-EM) reconstruction models,18,27 it has the highest resolution (3R0R: 2.35Å; 3JCI: 2.9Å; non-deposited density map: 4.5Å), which improves the atomic positions accuracy. The criteria of low energy folding were used to select the best model that was subsequently validated and used to build the capsid structure. Stereochemical quality and accuracy of the predicted model were evaluated using Ramachandran plot analysis.
The pentamer was generated by SYMMDOCK server28 and the generated models were visualized and compared to PCV2CS capsid protein using the PyMOL Version 1.3 (Schrodinger, LLC). Additionally, the model was submitted to the structure analysis at PDBSum server,29 in order to make structural inferences about the residues predicted to epitopes recognition.13 The capsid structure was built using icosahedral symmetry by Chimera 1.10.2 software.30
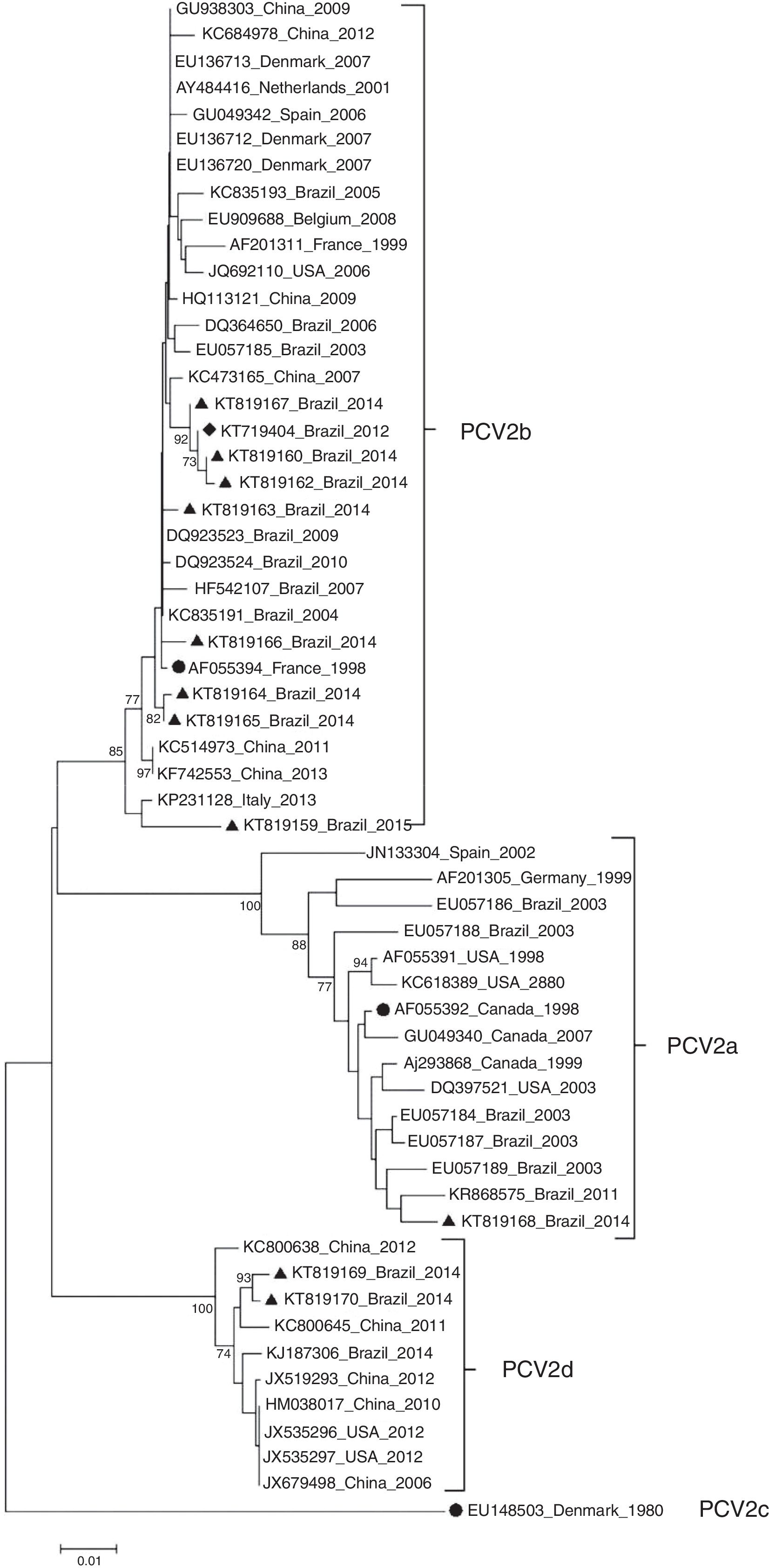
ResultsDNA sequencing and phylogenetic analysisBased on the phylogenetic tree reconstruction and on the identification of marker positions of the ORF2 gene suggested by Franzo et al.,31 two PCV2 genotypes were identified among the Brazilian PCV2 sequences: nine PCV2b (KT719404, KT819159, KT819160, KT819162, KT819163, KT819164, KT819165, KT819166, KT819167) and two PCV2d (KT819169 and KT819170) (Fig. 1).
, PCV2b (AF055394) and PCV2c (EU148503) are indicated by a solid circle (●). PCV2 (KT719404) is indicated by a solid diamond (♦) and the other 10 PCV2 isolates from PCVD cases that occurred in Brazil during 2014 and 2015 are indicated by a solid triangle (▴). The phylogenetic tree was constructed based on the nucleotide sequences of the ORF2 gene. The tree was inferred by using the Neighbor-Joining method with 1000 bootstrap replicates (Bootstrap values ≥70% are shown). The evolutionary distances were computed using the Maximum Composite Likelihood method, and are in the units of the number of base substitutions per site. Evolutionary analyses were conducted in MEGA6.")
Phylogenetic tree of 11 PCV2 in the present study and 47 PCV2 isolates representative of genotypes PCV2a, PCV2b, PCV2c and PCV2d. Reference strains from PCV2a (AF055392), PCV2b (AF055394) and PCV2c (EU148503) are indicated by a solid circle (●). PCV2 (KT719404) is indicated by a solid diamond (♦) and the other 10 PCV2 isolates from PCVD cases that occurred in Brazil during 2014 and 2015 are indicated by a solid triangle (▴). The phylogenetic tree was constructed based on the nucleotide sequences of the ORF2 gene. The tree was inferred by using the Neighbor-Joining method with 1000 bootstrap replicates (Bootstrap values ≥70% are shown). The evolutionary distances were computed using the Maximum Composite Likelihood method, and are in the units of the number of base substitutions per site. Evolutionary analyses were conducted in MEGA6.
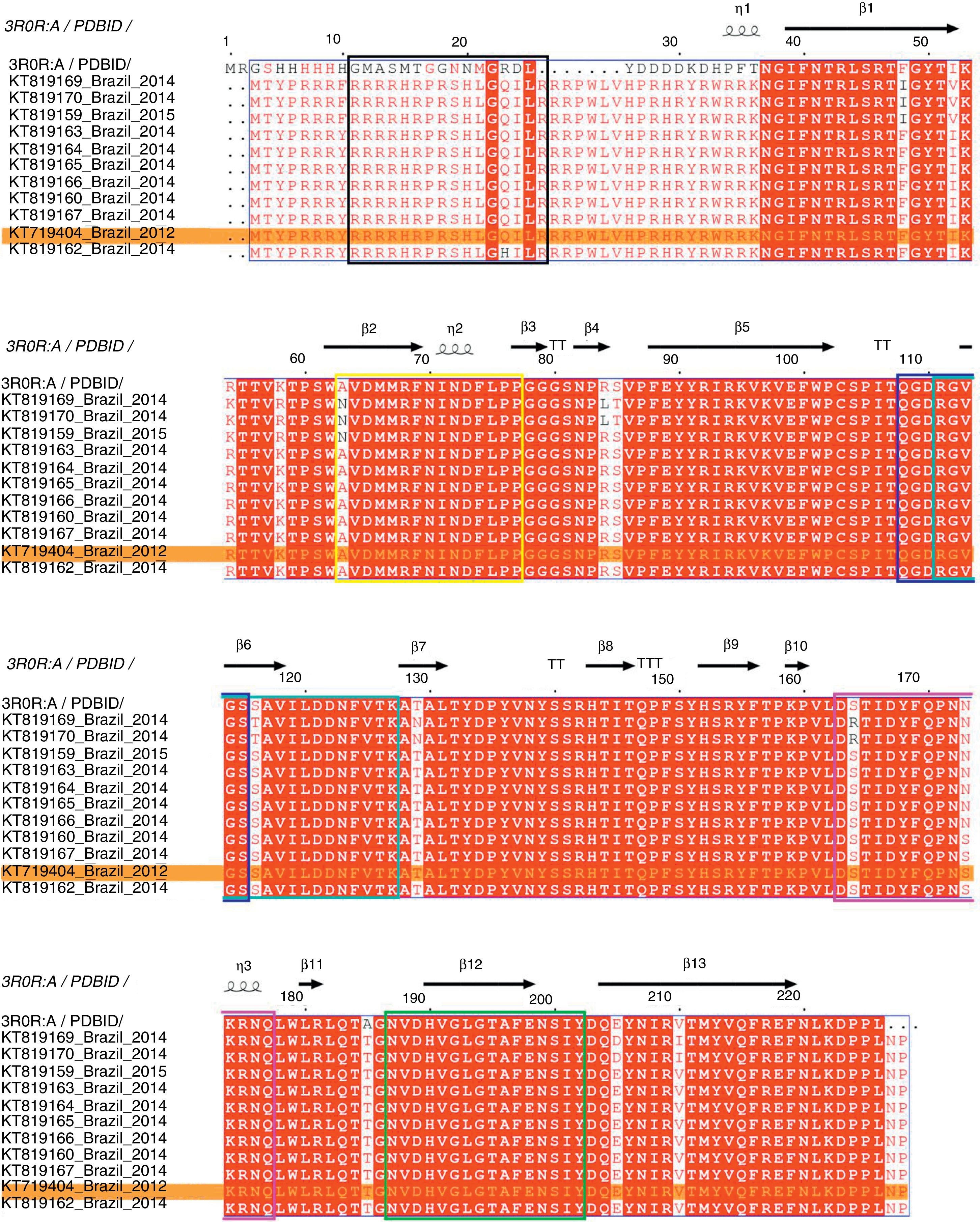
The complete genome sequence of all PCV2 isolates had 1767–1768 nucleotides of length and the ORF2 gene codified for a viral capsid protein with 233 amino acids. The amino acid sequence analysis of the capsid protein revealed three amino acid substitutions, in the positions 57 (F to I), 178 (N to S) and 190 (A to T). Moreover, the comparison between deduced amino acid sequences of all PCV2 isolates detected in PCV2-vaccinated farms showed that the amino acid threonine in position 190 was conserved in all PCV2 sequences analyzed. Fig. 2 represents the PCV2 sequences obtained in this study aligned with PCV2CS,13,25 where the relative numbering is displaced in −9 for F57I mutation and −5 for N178S and A190T mutations in comparison with PCV2CS sequence.
 in all analyzed Brazilian PCV2 sequences. The predicted antigenic regions, previously recognized as epitopes A to F,22 are demarcated by the colored boxes, in black, yellow, dark blue, cyan, pink and green, respectively. PCV2 (KT719404) is highlighted in an orange box.")
Multiple sequence alignment of the amino acid sequences of PCV2 capsid protein. Representation of conserved and variable residues in the capsid protein of 11 PCV2 isolates from PCVD cases that occurred in Brazil between 2012 and 2015 and the consensus sequence of the crystallographic structure of PDB. ID 3R0R:A. The diagram was generated using ESPript 3.0.13 The relative numbering is displaced in −9 for F57I mutation and −5 for N178S and A190T mutations in comparison with PCV2CS sequence. The alignment shows the residue conservation mainly at position 190 (185) in all analyzed Brazilian PCV2 sequences. The predicted antigenic regions, previously recognized as epitopes A to F,22 are demarcated by the colored boxes, in black, yellow, dark blue, cyan, pink and green, respectively. PCV2 (KT719404) is highlighted in an orange box.
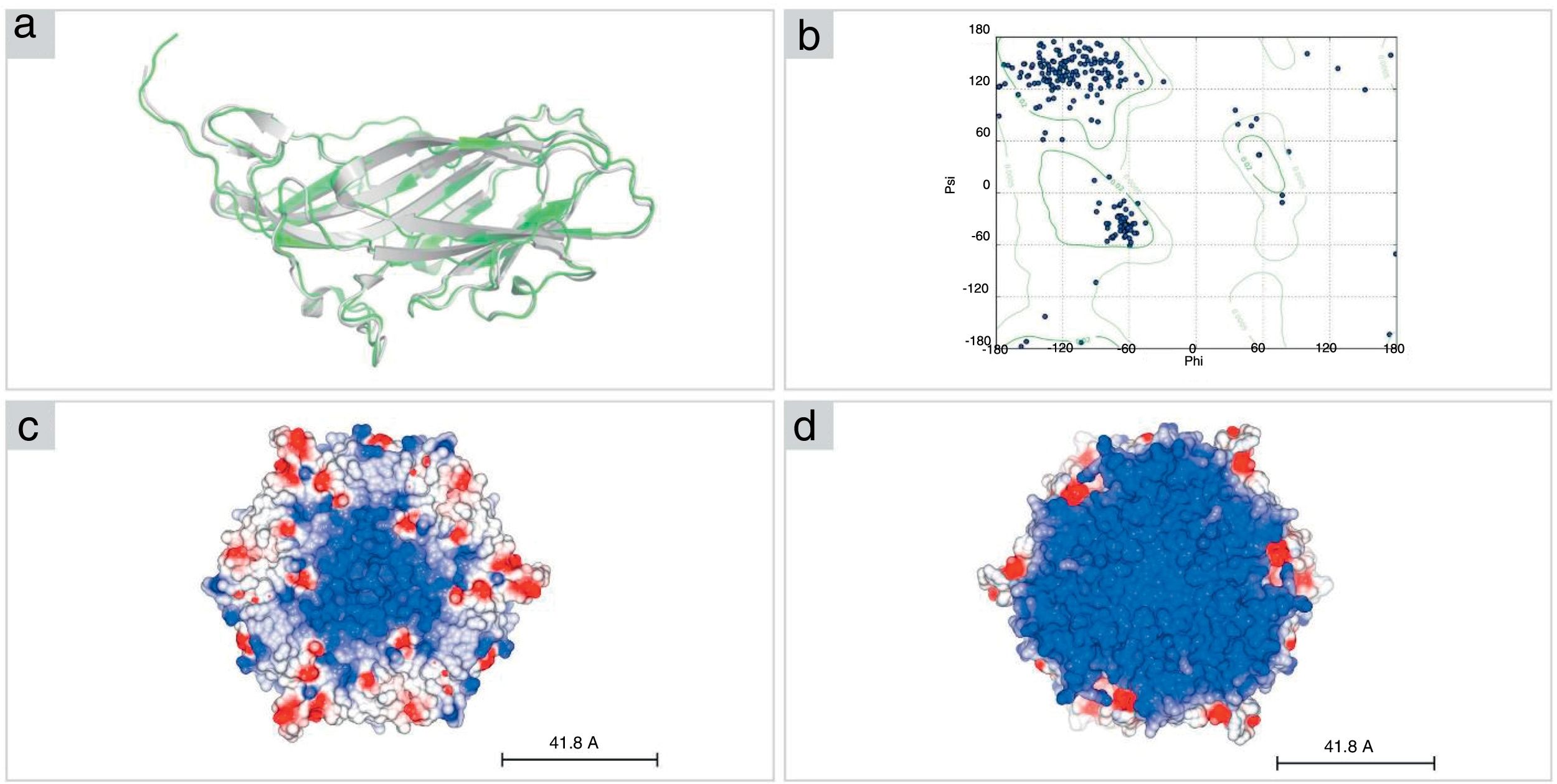
The structural model of PCV2 (KT719404) capsid protein, obtained by I-TASSER, presents a β-sandwich-like fold (a canonical viral jelly roll with eight β-strands organized in two sheets), analogous to PCV2 consensus sequence (PCV2CS) virus-like particle (VLP). PCV2CS structure13 and other structures previously determined by Cryo-EM18 or by homology modeling methods.16,17 The amino acid sequence of PCV2 (KT719404) presented 99% of identity when compared with PCV2CS. Furthermore, our model presented a root mean square deviation (RMSD) value of 0.962Å in comparison with the crystallographic structure (Fig. 3A). Additionally, analysis of the protein structure using the Ramachandran plot showed that 98.2% of amino acid residues from the structure modeled by I-TASSER were located in allowed regions of the plot (Fig. 3B), confirming the reliability of the proposed model.
 The structural model of PCV2 (KT719404; green) was built using the PDB.ID 3R0R:A crystal structure as a template (gray). (B) The Ramachandran plot shows 98% of residues in allowed regions. (C, D) Representation of pentamer subunits in electrostatic surface shows mostly positive surface (blue) in the interior (right) of the capsid in comparison with the exterior surface (left) of the capsid, where it is possible to visualize positive residues (red).")
Structural model of PCV2 capsid protein. (A) The structural model of PCV2 (KT719404; green) was built using the PDB.ID 3R0R:A crystal structure as a template (gray). (B) The Ramachandran plot shows 98% of residues in allowed regions. (C, D) Representation of pentamer subunits in electrostatic surface shows mostly positive surface (blue) in the interior (right) of the capsid in comparison with the exterior surface (left) of the capsid, where it is possible to visualize positive residues (red).
The capsid structure modeling revealed a trimer as the biological unity of PCV2 capsid protein obtained by 3-fold axes symmetry by SYMMDOCK. The concavity and the electrostatic potential surface of the pentamer indicates a predominantly positive region in the inner side of this complex (Fig. 3C), which is complementary with the nucleic acids that are composed mostly by negative charges, in comparison with the outer side (Fig. 3D).
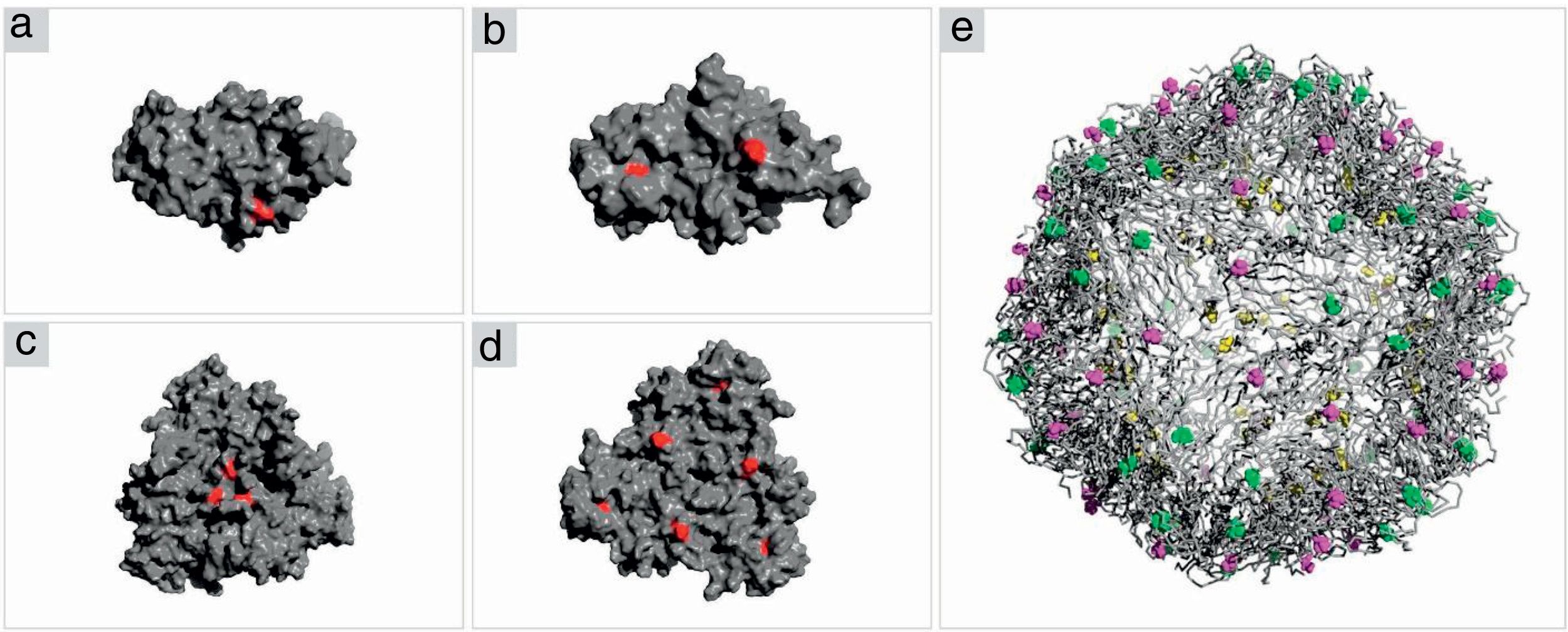
Structure analysis of the PCV2 capsid protein and epitope site recognitionThree distinct amino acid substitutions were detected in the PCV2 isolate (KT719404) at residues 57, 178 and 190 and were shown in the icosahedral capsid model (Fig. 4E). The phenylalanine to isoleucine (F57I) substitution results in a drastic change of the amino acid characteristic, but it does not provide an important alteration for the epitope recognition, since it is an internalized position (Fig. 4A–C). The amino acid substitutions asparagine to serine (N178S) and alanine to threonine (A190T) were observed in the generated model and might be associated with the predicted epitope site recognition due to their structural proximity.13 The N178S mutation is located at the predicted epitope D (Fig. 2), but facing into the interior of the viral capsid (Fig. 4B–D). In addition, the mutated residue (178) does not provide an abrupt change in the physical chemical characteristics of the residue. On the other hand, mutation A190T is located adjacent to the predicted epitope E, as shown in Fig. 2, and it is projected to the exterior surface of the capsid (Fig. 4C). Furthermore, the amino acid substitution A190T provides a slight and local physical–chemical change, from a nonpolar and hydrophobic side chain to a forked side chain with a hydroxyl group (Fig. 5A–C), changing the polar contacts, which is considered to be crucial to the antibody recognition.
 capsid protein. (A, B) Representation of the ORF2 monomer surface (gray) with amino acids changes F57I, N178S and A190T highlighted in red. (C, D) The trimeric subunits representing the changes mentioned above. (E) The icosahedral capsid model generated using ORF2 trimeric subunits. The mutated residues are highlighted in yellow (F57I), green (N178S) and purple (A190T).")
Crystal structure of PCV2 (KT719404) capsid protein. (A, B) Representation of the ORF2 monomer surface (gray) with amino acids changes F57I, N178S and A190T highlighted in red. (C, D) The trimeric subunits representing the changes mentioned above. (E) The icosahedral capsid model generated using ORF2 trimeric subunits. The mutated residues are highlighted in yellow (F57I), green (N178S) and purple (A190T).
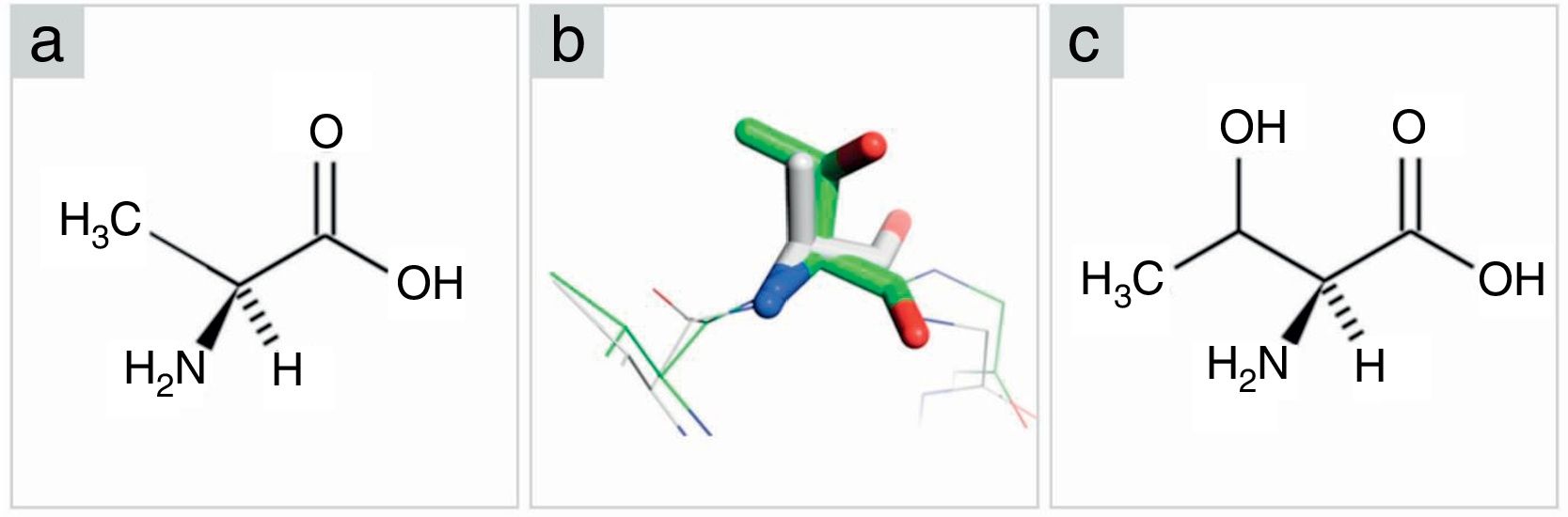
. (A) Alanine in Fischer projection. (B) Superposition between alanine from PCV2CS structure model (gray) and threonine from KT719404 sequence homology model (green). Oxygen and nitrogen are represented in red and blue, respectively. (C) Threonine in Fischer projection.")
Comparison among amino acids structure (alanine-threonine). (A) Alanine in Fischer projection. (B) Superposition between alanine from PCV2CS structure model (gray) and threonine from KT719404 sequence homology model (green). Oxygen and nitrogen are represented in red and blue, respectively. (C) Threonine in Fischer projection.
This is one of the first descriptions of PCV2-SD caused by a PCV2b genotype in vaccinated pig herds in Brazil. In the country, both PCV2a and PCV2b genotypes have been detected in pigs.32,33 Despite of the continuous use of vaccines over the last years in Brazil, since 2012, PCV2-SD cases have been reported in PCV2-vaccinated farms. These apparent vaccine failures may be a result of improper adherence to vaccination protocols or due to viral genetic diversity caused by selective pressure induced by vaccination.9 In this study, the molecular modeling methodology was applied to characterize a PCV2b strain isolated from a PCVD-SD case reported in a vaccinated pig farm in Brazil.
It has been shown that PCV2 has one of the highest rate of nucleotide substitution (approximately 1.2×10−3 substitutions/site/year) among the DNA viruses.34 Therefore, the occurrence of mutations in the capsid protein over time is expected. Indeed, in the present study, three amino acid changes (F57I, N178S and A190T) were observed in the capsid protein sequence of the PCV2.
To assess the occurrence of changes in the epitope conformation of the capsid protein, a molecular model was built using homology modeling based on the consensus sequence of PCV2 N-terminal truncated structure resolved by X-ray crystallography.13 As previously mentioned, the amino acid sequences of PCV2 isolate (KT719404) and PCV2CS had 99% of identity. Both sequences differed in the N-terminal region, where PCV2CS is added by a poly-His tag to allow the purification by affinity chromatography.13 However, the major part of sequence is conserved and reveals an internal jellyroll structure folding. Previous studies based on the PCV2CS crystallographic structure reported some predicted epitope recognition sites and suggested potential regions for antibody interaction.13,16,17,32,35 However, it is difficult to infer the precise location of the epitope recognition site based in a monomeric form, since many protein interfaces are occluded during the capsid assembly. Furthermore, protein structure analyses using incomplete macromolecular assemblies may not accurately reflect the residue accessibility to the solvent, which can lead to erroneous interpretation of the predicted epitope recognition sites. Consequently, structural model of the capsid protein obtained by experimental data or by in silico analyses is required to correctly infer the mutations or epitope site recognition spot. Therefore, in the present work, the icosahedral symmetry was applied to build a PCV2 capsid model where epitopes and mutation sites were mapped in the tridimensional structure.
Additionally, aiming to evaluate the possible role of the mutations found in PCV2 capsid protein in the reported cases of PCV2-SD in vaccinated pig herds in Brazil, the position of the three mutations (F57I, N178S and A190T) was identified in the PCV2 capsid model (KT719404). In spite of causing a drastic change in the amino acid characteristic, the mutation F57I did not provide an important alteration on the predicted epitope recognition, once it is an internalized position in the viral capsid. Similarly, the mutation N178S, although it is located in a previously predicted epitope recognition site (epitope D13), it is also internalized in the capsid structure. On the other hand, the single mutation A190T is exposed to the solvent and spatially close to the predicted epitope recognition pocket (epitope E13). Moreover, the change on the physicochemical properties of the residue 190 is most pronounced. Alanine is a non-polar residue with a small and hydrophobic side-chain and threonine is a bifurcate polar residue with a hydroxyl group in the side-chain. This mutation may change the epitope recognition pocket, which might hinder the interaction between antigen and antibody. Although, the amino acid position 190 has not been previously predicted as a key point for epitope recognition, recent publications7,35 have shown that changes at residue 190 were entailed in the antibody non-recognition. It is noteworthy to mention the difficulties in performing in vivo experiments, given the inability to reproduce PCV2-SD under experimental conditions.11 Because of this, in silico analysis by molecular modeling is a feasible approach to the understanding and interpretation of this complex system, as demonstrated in previous studies.16,17
In our hypothesis, the amino acid change from an alanine to a threonine at position 190 altered the capsid conformation within epitope E, from a non-polar hydrophobic residue to a polar configuration, which could modify the antigen-antibody interaction. Trible et al.15 determined that the region 161–207 of the capsid protein is responsible to the main difference on the immune response of vaccinated and PCV2-SD-affected pigs. In this sense, the level of protection conferred in subclinical infection or in clinical disease could be dependent on antibodies aimed against a single conformational epitope. Thus, it was suggested that the epitope located in this region could act as a decoy by focusing the antibody response on a nonprotective epitope, which allows the PCV2 to escape the humoral immunity. According to Krakowka et al.,36 although other variables might play a role in the PCV2-SD triggering, additional virulence determinants including multiple or even single amino acid changes in the capsid protein sequence could modify the spatial configuration of conformational epitopes, and therefore affect the PCV2 pathogenicity and recognition by neutralizing antibodies.37
Finally, the investigation of eleven PCV2-SD cases from eight distinct pig farms in Southern Brazil with a history of PCV2 vaccination failure revealed the circulation of PCV2d, besides PCV2b. Interestingly, the single mutation A190T is conserved in all PCV2b and PCV2d strains analyzed. In conclusion, the continuous monitoring for new PCV2 strains and identifying the biological importance of novel mutations will help to unravel the potential vaccine failures observed in field and to improve the current vaccines and vaccination programs.
Authors’ contributionsDG, VHBS, MEC and NM performed the analysis; DG, VHBS, LTF and RS wrote the manuscript; DG, VHBS, MEC, LTF, JRCZ and RS have revised the manuscript and cooperate for its improvement. All authors read and approved the final manuscript.
Conflicts of interestAuthors declare that they have no conflicts of interest.
We thank Neide L. Simon for the technical assistance on DNA sequencing. The authors sincerely appreciate the technical assistance of Marina Schmitt for the graphic design. This study was funded by the Brazilian Agricultural Research Corporation-EMBRAPA (02.11.01.006.00) and partially supported by the São Paulo Research Foundation-FAPESP (grant 12/23730-1) and CAPES grant (VHBS). JRCZ is a fellow of CNPq.