








Wastewater from an anaerobic treatment plant at a slaughterhouse was analysed to determine the bacterial biodiversity present. Molecular analysis of the anaerobic sludge obtained from the treatment plant showed significant diversity, as 27 different phyla were identified. Firmicutes, Proteobacteria, Bacteroidetes, Thermotogae, Euryarchaeota (methanogens), and msbl6 (candidate division) were the dominant phyla of the anaerobic treatment plant and represented 21.7%, 18.5%, 11.5%, 9.4%, 8.9%, and 8.8% of the total bacteria identified, respectively. The dominant bacteria isolated were Clostridium, Bacteroides, Desulfobulbus, Desulfomicrobium, Desulfovibrio and Desulfotomaculum. Our results revealed the presence of new species, genera and families of microorganisms. The most interesting strains were characterised. Three new bacteria involved in anaerobic digestion of abattoir wastewater were published.
For hygienic reasons, abattoirs use copious amounts of water in their processing operations (slaughtering and cleaning), which creates significant wastewater. In addition, the increased use of automated machines to process carcasses, along with the incorporation of washing at every stage, has increased water consumption in slaughterhouse facilities. The high fat and protein content of slaughterhouse waste makes wastewater a good substrate for anaerobic digestion, due to its expected high methane yield.1 Numerous microorganisms are involved in the anaerobic degradation of slaughterhouse waste, any step of which may be rate-limiting depending on the waste being treated as well as the process involved.
The microorganisms involved in anaerobic digestion have not been fully identified; however, at least four groups of microorganisms are involved in this process.2 The first group are the hydrolytic bacteria that degrade complex compounds (protein, carbohydrates, and fat) into simpler compounds, such as organic acids, alcohols, carbon dioxide (CO2) and hydrogen. The second group are the hydrogen producing acetogenic bacteria that use organic acids and alcohols to produce acetate and hydrogen. The third group contains homoacetogenic bacteria that can only form acetate from hydrogen, CO2, organic acids, alcohols, and carbohydrates. The fourth group comprises methanogens that form methane from acetate, CO2, and hydrogen. Hydrolytic, acetogenic, and methanogenic microorganisms play an equally important role in anaerobic digestion and methane production. Optimal methane production is only achieved via the interaction of multiple microorganisms,3 and therefore, biodegradation of molecules in wastewater depends on the activity of all microbial groups involved.
Common fermentative bacteria include Lactobacillus, Eubacterium, Clostridium, Escherichia coli, Fusobacterium, Bacteroides, Leuconostoc, and Klebsiella. Acetogenic bacteria include Acetobacterium, Clostridium, and Desulfovibrio.2 Methane producing organisms are classified under domain Archaea and phylum Euryarchaeota.4
In order to better understand the function of a bacterial population, a detailed description of the microbial ecosystem is necessary. One method is via molecular biology techniques.5 Recent advances in the molecular analysis of bacterial ecosystems allow a better understanding of the specific microorganisms involved in wastewater treatment. There are only a few studies focused on microbial populations, diversity and evolution in reactors fed with complex organic wastes.1 Therefore, little is known about the composition of these reactors. The development of advanced molecular biology techniques has contributed to the detection, quantification, and identification of the bacterial populations involved in the treatment of abattoir wastewater. For example, cloning and sequencing of 16S rRNA gene fragments provide information about the phylogeny of the microorganisms. Additionally, single stranded conformation polymorphism (SSCP) offers a simple, inexpensive and sensitive method for detecting whether or not DNA fragments are identical in sequence, and can greatly reduce the amount of sequencing necessary.6
This work aimed to study the bacterial ecology of an anaerobic digestor through both bacterial culture and molecular biological techniques. The bacteria involved in the anaerobic digestion of abattoir wastewater were identified using classic microbiology techniques and molecular tools (sequencing of 16S rRNA and SSCP). Additionally, our results were compared with those of Gannoun et al.6 to evaluate the effect of storage at 4°C on the bacterial diversity of the sludge.
Material and methodsOrigin of the sludgeAnaerobic sludge samples were collected from an upflow anaerobic filter that treats abattoir wastewater in Tunisia.7 The digestor operated under both mesophilic (37°C) and thermophilic (55°C) conditions. Samples were taken at the end of thermophilic phase and stored at 4°C. The sludge was then analysed to determine the bacterial diversity present, first via bacterial culture.
DNA extraction, PCR and SSCP analysis of the digestor sludgeFour milliliters of the anaerobic sludge sample were centrifuged at 6000rpm for 10min. Pellets were re-suspended in 4mL of 4M guanidine thiocyanate–0.1M Tris pH 7.5 and 600μL of N-lauroyl sarcosine 10%. Two hundred and fifty microlitres of treated samples were transferred in 2mL tubes and stored at −20°C.
Extraction and purification of total genomic DNA was implemented according to the protocol developed by Godon et al.8
Highly variable V3 regions of bacterial 16S rRNA genes were amplified by PCR using bacterial (w49–w34) primers (Table 1). Samples were treated according to the protocol previously described by Delbès et al.9
For electrophoresis, PCR–SSCP products were diluted in water before mixing with 18.75μL formamide (Genescan-Applied Biosystems) and 0.25μL internal standards (ROX, Genescan-Applied Biosystems) according to the protocol of SSCP described by Delbès et al.9
SSCP analyses were performed on an automatic sequencer abi310 (Applied Biosystems). RNA fragment detection was done on the fluorescent w34 primer. The results obtained were analysed by GeneScan Analysis 2.0.2 Software (Applied Biosystems) as specified by Gannoun et al.6 For bacterial identification, pyrosequencing of the DNA samples using a 454 protocol was performed (Research and Testing Laboratory, Lubbock, USA).
Methods of analysis for pyrosequencing data used herein have been described previously.10–14 Sequences are first depleted of barcodes and primers. Then, short sequences under 200bp are removed, sequences with ambiguous base calls are removed, and sequences with homopolymer runs exceeding 6bp are removed. Sequences are then denoised and chimeras removed. Operational taxonomic units were defined after removal of singleton sequences, clustering at 3% divergence (97% similarity).15–21 Operational taxonomic units were then taxonomically classified using BLASTn against a curated GreenGenes database22 and compiled into each taxonomic level into both “counts” and “percentage” files. Counts files contain the actual number of sequences, while the percentage files contain the percentage of sequences within each sample that map to the designated taxonomic classification.
Enrichment and isolation procedures of fermentative and SRB bacteriaThe Hungate technique23 was used throughout this study. Inoculations were done with 10% of culture. Samples were collected from the anaerobic filter at the end of thermophilic phase. A 0.5mL aliquot of sample was inoculated into Hungate tubes containing 5mL of basal medium.
For both SRB and fermentative bacteria, enrichment and isolation was done according to the protocol described previously by Hungate23 and Khelifi et al.5
Purification of the DNA, PCR amplification and sequencing of the 16S rRNA gene of isolated bacteriaPurification of the DNA, PCR amplification and sequencing of the 16S rRNA gene were performed as described previously.24 The partial sequences generated were assembled using BioEdit version 5.0.925 and the consensus sequence of 1495 nucleotides was corrected manually for errors. The most closely related sequences in GenBank (version 178), the Ribosomal Database Project (release 10) identified using BLAST,26 and the Sequence Match program,27 were extracted and aligned. The consensus sequence was then adjusted manually to conform to the 16S rRNA secondary structure model.28 Nucleotide ambiguities were omitted and evolutionary distances were calculated using the Jukes and Cantor option.29 Dendrograms were constructed with the TREECON program using the neighbor-joining method.30 Tree topology was re-examined by the bootstrap method (1000 replications) of re-sampling.31 Its topology was also supported using the maximum-parsimony and maximum-likelihood algorithms.
Data analysesThe two main factors taken into account when measuring diversity are richness and evenness. Richness is a measure of the number of different kinds of organisms present in a particular sample. Evenness (Es) compares the similarity of population sizes between each of the species present. The reciprocal of Simpson's index (diversity richness) (1/D), which is widely used for ecological studies, was also used as a measure of diversity. Richness and evenness were calculated and interpreted as described previously by Simpson32 and Gannoun et al.6
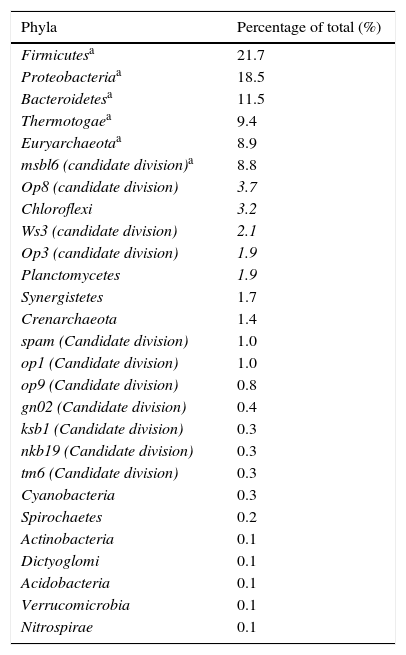
Results and discussionDiversity and abundance of the bacterial communities in the bioreactor sludge using SSCP and DNA sequencingThere was significant microbial diversity of the upflow anaerobic filter, which operated under both mesophilic (37°C) and thermophilic (55°C) conditions. Twenty-seven different phyla were identified, and the six most common phyla (Firmicutes, Proteobacteria, Bacteroidetes, Thermotogae, Euryarchaeota (the methanogenic bacteria), and the msbl6 (candidate division)) represented 78.8% of the total (Table 2). The 21 less common phyla represented 21% of the total.
The different phyla found in the digestor.
| Phyla | Percentage of total (%) |
|---|---|
| Firmicutesa | 21.7 |
| Proteobacteriaa | 18.5 |
| Bacteroidetesa | 11.5 |
| Thermotogaea | 9.4 |
| Euryarchaeotaa | 8.9 |
| msbl6 (candidate division)a | 8.8 |
| Op8 (candidate division) | 3.7 |
| Chloroflexi | 3.2 |
| Ws3 (candidate division) | 2.1 |
| Op3 (candidate division) | 1.9 |
| Planctomycetes | 1.9 |
| Synergistetes | 1.7 |
| Crenarchaeota | 1.4 |
| spam (Candidate division) | 1.0 |
| op1 (Candidate division) | 1.0 |
| op9 (Candidate division) | 0.8 |
| gn02 (Candidate division) | 0.4 |
| ksb1 (Candidate division) | 0.3 |
| nkb19 (Candidate division) | 0.3 |
| tm6 (Candidate division) | 0.3 |
| Cyanobacteria | 0.3 |
| Spirochaetes | 0.2 |
| Actinobacteria | 0.1 |
| Dictyoglomi | 0.1 |
| Acidobacteria | 0.1 |
| Verrucomicrobia | 0.1 |
| Nitrospirae | 0.1 |
Gram-positive bacteria, including Firmicutes (low G+C), were the most common type of bacteria in the anaerobic digestor, comprising 21.7% of the total. Both Gram-positive low G+C bacteria and the Bacteroidetes phylum are known for their fermentative properties. Furthermore Bacteroidetes play an important role in the degradation of complex polymers. Firmicutes and Bacteroidetes are also the two main groups encountered in the study by Godon et al.8 of a fluidised bed anaerobic digestor fed with vinasse. These two groups of bacteria hydrolyse the polymer substrates which are not degraded during the previous stages of anaerobic digestion (such as polysaccharides, proteins and lipids) into acetate, long chain fatty acids, CO2, formate and hydrogen.
Bacteria within the Proteobacteria phylum were also commonly found in the digestor. These Gram-negative bacteria are considered to be some of the most cultivable microorganisms .33,34 The Proteobacteria have an important role in the hydrolysis and acetogenesis steps of anaerobic digestion, and include delta, gamma and beta varieties. Deltaproteobacteria contains many syntrophic anaerobic bacteria, which participate in sulphate reduction. Among the Gammaproteobacteria, there are many denitrifying bacteria or bacteria that accumulate phosphates .35 The Betaproteobacteria are involved in nitrification, and are potentially also involved in denitrification.
Phylogenetic analysis of the domain Bacteria also helped to highlight the existence of a poorly known order, Thermotogales, which was relatively abundant within the digestor at 9.4%. Thermotogales contains anaerobic bacteria that are heterotrophic with a fermentative metabolism.36 These bacteria are also found in other anaerobic digestors.8
The Planctomycetales group represented 1.9% of the bacteria within the digestor. Bacteria within Planctomycetales are limited to five kinds and only eight species are described. Aerobic heterotrophic Planctomycetes have been successfully isolated from brackish marine sediments, freshwater sediments, soil, hot springs, salt pits and tissues from giant tiger prawn postlarvae.37,38 In addition, a special group of Planctomycetes were implicated in the oxidation of ammonia under anaerobic conditions in wastewater plants, coastal marine sediments, and oceanic and freshwater oxygen minimum zones.39 Furthermore, a wide variety of Planctomycetales were found during analysis of samples from aquatic anaerobic environments, a sulphide- and sulphur-rich spring, activated sludge wastewater treatment plants and in anaerobic digestors.8,38,39
The Chloroflexi represented 3.2% of the digestor's bacteria. Chloroflexi have been identified from many environments through 16S rRNA gene profiling, including marine and freshwater sediments. Despite this, the Chloroflexi remain a relatively understudied bacterial lineage. At present, there are 19 complete genomes available for the Chloroflexi.40
Tang et al.41 showed that three phyla were involved in the mesophilic anaerobic treatment of synthetic rejection containing bovine serum albumin: Bacteroidetes, followed by Firmicutes and Proteobacteria. In another study by Fang et al.42 that evaluated the anaerobic degradation of phenol rich rejection in an upflow anaerobic sludge blanket (UASB) reactor, eight phylogenetic groups were detected, namely Thermotogales (38.9% of clones), Firmicutes (27.8%), Chloroflexi (11.1%), candidate division OP8 (9.3%), candidate division OP 5 (5.5%), Proteobacteria (3.7%), Bacteroidetes (1.9%) and Nitrospirae (1.9%). These results are comparable with the results of our study.
The Euryarchaeota, which consist mainly of methanogenic bacteria, represented 8.9% of bacteria within the anaerobic digestor. The Crenarchaeota, extreme thermoacidophiles, were also detected in the digestor with an abundance of 1.4%. The digestor seems to have limited archaeal diversity. These results are similar to the results of another study on the bacterial diversity of an anaerobic digestor fed with vinasse, which contained only 4% of Archaea Bacteria sequences.8
Overall, there was significant bacterial diversity within the digestor. There were 23 species each consisting of greater than 1%, and the most abundant species was Kosmotoga spp. with a percentage of 9.37% (Table 3).
The main genus and species of the digestor.
| Strain | Percentage (%) | Strain | Percentage (%) |
|---|---|---|---|
| Kosmotoga spp.a | 9.37 | Cclostridium acetireducens | 0.97 |
| msbl6 (candidate division)a | 8.75 | Segetibacter spp. | 0.97 |
| Desulfotomaculum thermocisternuma | 6.25 | Clostridium limosum | 0.90 |
| Rriemerella anatipestifera | 5.48 | Sulfurovum lithotrophicum | 0.90 |
| Pelotomaculum thermopropionicuma | 4.44 | op9 (candidate division) | 0.83 |
| op8 (candidate division)a | 3.68 | Achromatium oxaliferum | 0.83 |
| Tthiohalomonas denitrificansa | 3.12 | Ureibacillus suwonensis | 0.76 |
| Thermobaculum terrenuma | 3.12 | Desulfobacterium catecholicum | 0.76 |
| Desulfobulbus elongatusa | 2.63 | Synergistes spp. | 0.76 |
| Methanobacterium spp.a | 2.43 | Desulfomicrobium spp. | 0.76 |
| Dysgonomonas mossiia | 2.22 | Desulfobulbus spp. | 0.69 |
| Caldanaerobacter uzonensisa | 2.15 | Methanobacterium beijingense | 0.62 |
| ws3 (candidate division)a | 2.08 | Pseudomonas resinovorans | 0.62 |
| op3 (candidate division)a | 1.94 | Rhodovibrio salinarum | 0.55 |
| Pirellula spp.a | 1.66 | Legionella birminghamiensis | 0.55 |
| Methanobacterium aarhusensea | 1.45 | Methanosaeta thermophila | 0.55 |
| Nitrosococcus oceania | 1.45 | Acetobacterium wieringae | 0.55 |
| Coprococcus clostridium sp. ss2/1a | 1.38 | Thermanaerovibrio acidaminovorans | 0.55 |
| Methanosphaerula palustrisa | 1.11 | Thermosinus carboxydivorans | 0.55 |
| Parabacteroides goldsteiniia | 1.04 | Ruminococcus flavefaciens | 0.48 |
| Candidatus nitrososphaera gargensisa | 1.04 | Methanosaeta spp. | 0.48 |
| spam (candidate division)a | 1.04 | Rhodovulum euryhalinum | 0.48 |
| op1 (candidate division)a | 1.04 | Thermomonas fusca | 0.48 |
| Methanobrevibacter curvatus | 0.97 | Mechercharimyces mesophilus | 0.41 |
The 48 most abundant species in the sample.
SSCP analyses (Fig. 1) show the results of two samples of sludge collected from the same digestor at the end of the thermophilic phase.6 The second sample was stored at 4°C for two months and showed different SSCP patterns. The analysis of the two SSCP patterns showed significant change in the bacterial community over time, which can be explained by the fact the sludge is not stable over time.

The dynamics of bacterial communities were monitored by PCR-SSCP methods. The profile obtained for the domain Bacteria is shown in Fig. 1. The SSCP pattern revealed the high diversity of bacteria, with at least 48 distinguishable peaks and about 23 prominent peaks. The bacterial diversity richness (1/D) and species evenness (Es) were used as a measure of diversity and abundance. The obtained values were 38.97 (which offers toward the number of species S=48) (indicated maximum diversity)) and 0.811 (which offers toward to 1: species in the sample are quite evenly distributed), respectively. This result is in agreement with other molecular studies based on the PCR-SSCP methods.
Several research groups confirmed our results and demonstrated that diversity in bacterial digestors varies depending on several factors. However, Zumstein et al.,43 who studied the community dynamics in an anaerobic bioreactor using fluorescence PCR single-strand conformation polymorphism analysis, indicated that throughout the period of the study, rapid significant shifts in the species composition of the bacterial community occurred. In fact, the bacterial community was followed for two years through the analysis of 13 SSCP patterns, with one SSCP pattern every two months. The analysis of the SSCP patterns showed a continuous change in the bacterial community during that time. Typically, a microorganism initially present at low levels in the community grew, peaked and decreased. Moreover, some microorganisms seemed to fluctuate simultaneously.
Keskes et al.44 studied the effect of the prolonged sludge retention time on bacterial communities involved in the aerobic treatment of abattoir wastewater by a submerged membrane bioreactor. Their results showed that the biodiversity varied significantly in relation with the environmental conditions, particularly TSS.
The sludge microorganisms and associations of microorganisms may occupy the same ecological niche successively. They could correspond to the ecological unit called an ecotype.45
Isolation and identification of bacteriaFermentative bacteria and SRB involved in the anaerobic digestion of organic matter in abattoir effluents were investigated by two approaches: classical microbiology and molecular taxonomy.
The isolation of bacteria is preceded by an enrichment phase which favors the growth of a given microorganism (fermentative bacteria or SRB), selected according to the physical and nutritional conditions of the medium category. For this purpose, two different culture media were used, as described in the Materials and Methods section. The first culture medium was specially designed for the detection of fermentative bacteria and the second for SRB. Both culture media were tested at 37°C and 55°C. Glucose was used as an energy source to isolate fermentative bacteria, whereas lactate, acetate and H2CO2 were reserved for the isolation of SRB.
Isolation and culture of bacteria allowed us to study the biodiversity of these bioreactors as well as highlighted the dominant bacteria in the different conditions tested. The isolation of the different strains was carried out on an agar medium. This step helped to isolate strains that are identified based on their morphological differences (size, shape, and color).
Following this step, molecular analysis of the isolated strains was performed. After extraction of the bacterial DNA, the 16S rDNA was amplified by PCR. The quality of the amplified DNA was visualised with ultra-violet light after migration on gel agarose at 1%. The PCR product was subsequently sequenced to identify the different strains. The genes coding for 16S rRNA have been chosen as taxonomic markers, as they have a universal distribution and conserved function. These genes possess both highly conserved areas, providing information on the evolution of distant species, and variable areas, to differentiate organisms belonging to the same genus, and eventually the same species.46
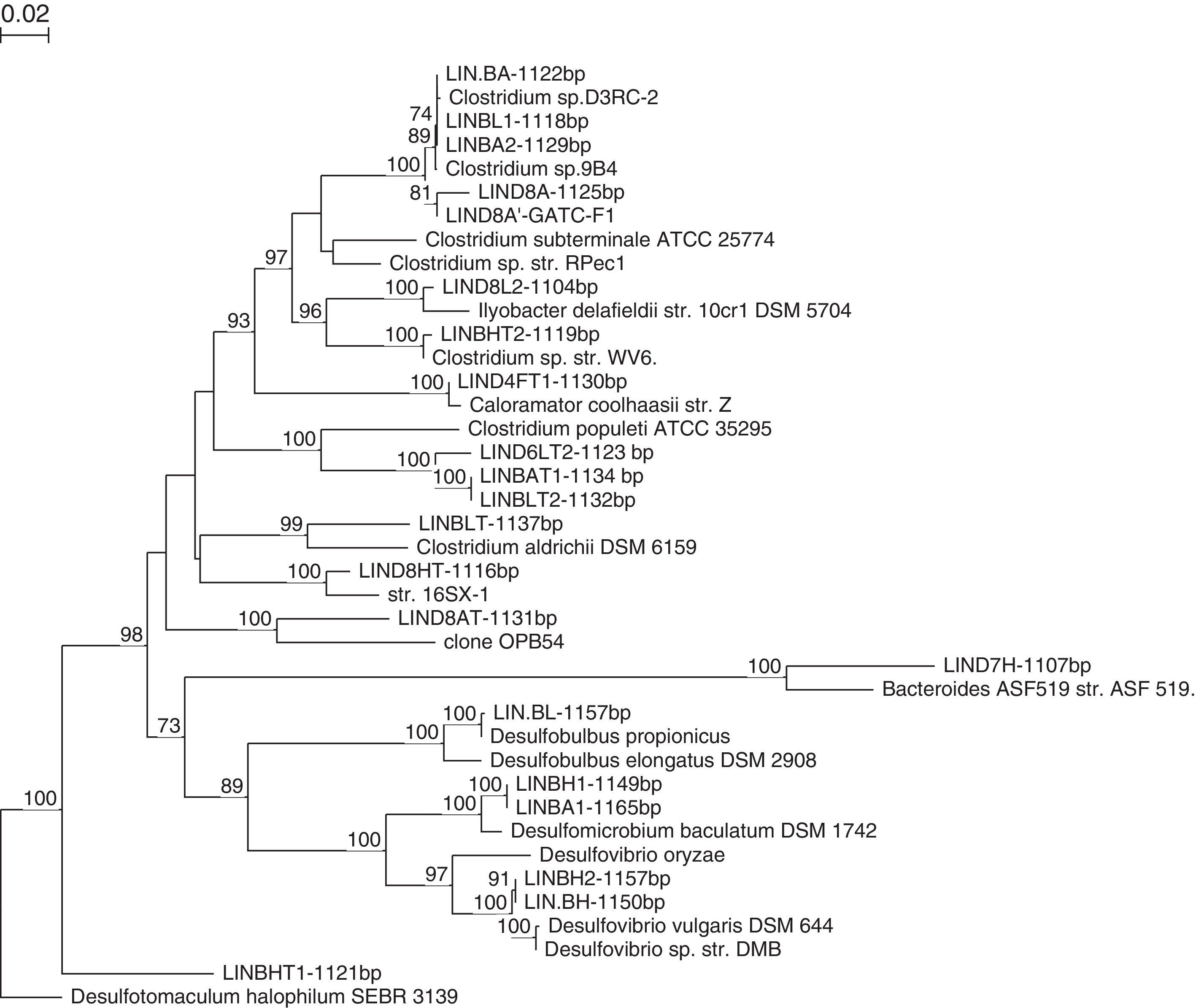
Each microorganism was then identified by comparing the 16S rDNA sequences with those of known microorganisms, and each was cataloged in computer databases. Given the large number of isolated bacteria in recent underwent restriction analysis by ARDRA technique to eliminate identical strains restriction profiles to keep only the amplifias of different strains priori which will be the object of phylogenetic studies. Phylogenetic affiliations of the 16S rRNA of bacteria sequences are presented in Table 4 with the closest relatives of isolated mesophilic and thermophilic fermentative bacteria and SRB.
Phylogenetic affiliation of the 16S rRNA bacteria sequences of isolated strains.
| Name | Closest neighbor | Accession number | Origin | References | Similarity (%) |
|---|---|---|---|---|---|
| Mesophilic fermentative bacteria (37°C) | |||||
| LIND8L2 | Clostridium novyi | AB041865 | Soil and feces | Sasaki et al.49 | 96% |
| LIND7Ha | Parabacteroides merdae | AB238928 | Human feces | Johnson et al.51 | 91% |
| LIND8A | Clostridium sp 13A1 | AY554421 | Anaerobic bioreactor treating cellulose waste | Burrell et al.50 | 99% |
| LINBA | Clostridium sp D3RC-2 | DQ852338 | Rumen yak china | Zhang et al.48 | 99% |
| LINBL1 | Clostridium sp D3RC-2 | DQ852338 | Rumen yak china | Zhang et al.48 | 99% |
| LINBA2 | Clostridium sp D3RC-2 | DQ852338 | Rumen yak china | Zhang et al.48 | 99% |
| LIND8A | Clostridium sp D3RC-2 | DQ852338 | Rumen yak china | Zhang et al.48 | 96% |
| Thermophilic fermentative bacteria (55°C) | |||||
| LIND6LT2a | Parasporobacterium paucivorans | AJ272036 | Anaerobic digestor treating solid waste | Lomans et al.57 | 87% |
| LIND8AT | Uncultured bacterium | DQ125705 | Soils contaminated with uranium waste | Brodie et al.59 | 97% |
| LINBAT1 | Uncultured bacterium | AF280825 | Anaerobic digestor treating pharmaceutical wastes | Lapara et al.60 | 99% |
| LINBLT2 | Uncultured bacterium | AF280825 | Anaerobic digestor treating pharmaceutical wastes | Lapara et al.60 | 98% |
| LIND4FT1 | Caloramator coolhaasii | AF104215 | Anaerobic thermophilic granular sludge | Plugge et al.61 | 96% |
| LIND8HT | Lutispora thermophila | AB186360 | Thermophilic bioreactor digesting municipal solid wastes | Shiratori et al.62 | 99% |
| LINBLT | Clostridium thermosuccinogenes | Y18180 | Cattle manure, beet pulp, soil, sediment pond | Stackebrandt et al.72 | 98% |
| LINBHT2 | Clostridium tertium | Y18174 | Open war wounds | Stackebrandt et al.72 | 98% |
| Mesophilic sulphate-reducing bacteria (37°C) | |||||
| LINBL | Desulfobulbus propionicus | AY548789 | Fluidized-bed reactors treating acidic, metal-containing wastewater | Kaksonen67 | 99% |
| LINBH | Desulfovibrio vulgaris | AE017285 | Soil, animal intestines and feces, fresh and salt water | Heidelberg et al.68 | 99% |
| LINBH2 | Desulfovibrio vulgaris | AE017285 | Soil, animal intestines and feces, fresh and salt water | Heidelberg et al.68 | 99% |
| LINBA1 | Desulfomicrobium baculatum | AJ277895 | Water-saturated manganese carbonate ore | Hippe73 | 99% |
| LINBH1 | Desulfomicrobium baculatum | AJ277895 | Water-saturated manganese carbonate ore | Hippe73 | 99% |
| Thermophilic sulphate-reducing bacteria (55°C) | |||||
| LINBHT1a | Desulfotomaculum halophilum | U88891 | Oil production facilities | Tardy-Jacquenod et al.69 | 89% |
Strains LINBA, LINBL1, LINBA2 are closest phylogenetically to Clostridium sp. D3RC-2 with a percentage sequence similarity of 99%. This bacterium was firstly detected in the rumen of a yak in China47,48 but it is not yet described. The strain LIND8A shares 96% of sequences with Clostridium sp. D3RC-2. This strain seems to be a new species and differs from the latter. The strain LIND8L2 has also been recently affiliated with Clostridia species. LIND8L2 is a strain similar to Clostridium novyi with 96% sequence similarity. C. novyi is a pathogenic bacterium phylogenetically close to C. botulinum and C. haemolyticum.49 The nearest strain to LIND8A is Clostridium sp 13A1, previously isolated from an anaerobic bioreactor treating cellulose wastes,50 with 99% sequence similarity. The closest phylogenetic relatives of the strain LIND7HT are Parabacteroides merdae,51P. goldsteinii52,53 and P. gordonii,54 with 91.4%, 91.3% and 91.2% sequence similarity, respectively. This novel strain was identified and characterised by Jabari et al.55 On the basis of phylogenetic inference and phenotypic properties, LIND7HT is proposed as the type strain of a novel genus and species within the family Porphyromonadaceae, Macellibacteroides fermentans gen. nov., sp. nov.
Strains isolated in mesophilic conditions were determined to belong to Firmicutes and Bacteroidetes, which are known for their fermentative activity and which are the two groups mainly encountered in the study by Godon et al.8 on an anaerobic digestor. They hydrolyse the polymer substrates not degraded during the stages of remediation (such as polysaccharides, proteins and lipids) to acetate, long chain fatty acids, CO2, formate and hydrogen.
The isolation of fermentative, thermophilic bacteria obtained some strains classified as “uncultured bacterium” indicating they cannot be grown on synthetic material in a laboratory. These strains are LIND6LT2, LIND8AT, LINBAT1 and LINBLT2. The strain LIND6LT2 was detected in an anaerobic digestor treating solid waste in thermophilic conditions.56 The closest phylogenetic relative to this strain is Parasporobacterium paucivorans with 87.17% sequence similarity.57 This novel strain was initially identified and characterised by Jabari et al.58 On the basis of phylogenetical and physiological properties, the strain LIND6LT2T is proposed as the strain type of Defluviitalea saccharophila gen. nov., sp. nov., placed in Defluviitaleaceae fam. nov., within the phylum Firmicutes, class Clostridia, order Clostridiales. The strain LIND8AT has 97% similarity with a strain classified as uncultivable bacterium, probably involved in metal reduction.59
According to the phylogenetic tree (Fig. 2), the strains LINBAT1 and LINBLT2 are phylogenetically close. These sequences were also identified within an anaerobic digestor treating pharmaceutical wastes.60 The closest relative to strain LIND4FT1 is Caloramator coolhaasii, with 96% sequence similarity which suggests that this strain is a new species. C. coolhaasii has been isolated from an anaerobic granular sludge bioreactor that degrades glutamate. It is moderately thermophilic and strictly anaerobic.61 LIND8HT strain is close to Lutispora thermophile,62 with 99% sequence similarity which is firstly isolated from an enrichment culture derived from an anaerobic thermophilic bioreactor treating artificial solid wastes. Strains LINBLT and LINBHT2 were affiliated with C. tertium and C. thermosuccinogenes, respectively, each with 98% sequence similarity.

The mesophilic and thermophilic fermentative strains which were isolated were also designated to the group Firmicutes, which seems to be the most abundant group in our anaerobic digestor. Almost all isolates were represented by Clostridium species. This is not surprising, because during the hydrolysis phase in bioreactors, macromolecules such as polysaccharides, lipids, proteins and nucleic acids are cleaved, typically by specific extracellular enzymes, producing monomers (monosaccharides, fatty acids, amino acids and nitrogen bases) which are transported into the cell where they are fermented. The bacteria involved in this stage have a strictly anaerobic or facultative metabolism. During the acidogenesis phase, these monomers are metabolised by fermentative microorganisms to primarily produce volatile fatty acids (acetate, propionate, butyrate, isobutyrate, valerate and isovalerate), but also alcohols, sulphide (H2S), CO2 and hydrogen. Acidogenesis leads to simplified products of fermentation, and the bacteria involved in this step may be facultative or strictly anaerobic. Strictly anaerobic bacteria of the genus Clostridium are often a large fraction of the population participating in the anaerobic step of acid formation.
Fermentative bacteria were abundant, particularly the proteolytic Clostridium species. These species hydrolyse proteins to polypeptides and amino acids, while lipids are hydrolysed via oxidation to long-chain fatty acids, and glycerol and polycarbohydrates are hydrolysed to sugars and alcohols. After that, fermentative bacteria convert the intermediates to volatile fatty acids, hydrogen and CO2.6
Sulphate-reducing bacterial communities in the sludge bioreactorSulphate-reducing bacteria were isolated in mesophilic and thermophilic conditions on various substrates. In anaerobic digestion, the acidogenesis products are converted into acetate and hydrogen in the acetogenesis phase. The hydrogen is normally used by the microbial community's methanogenic hydrogenophiles to reduce CO2 to methane (CH4) while acetate is converted by methanogenic acetoclastes to CH4.
The presence of sulphate in the medium may change the flow of the substrate available for methanogens. In fact, the SRB may oxidise a portion of the substrate (mainly via the hydrogen) using sulphate as an electron acceptor. In such situations, the substrate is converted to sulphur if the pH of the medium is acidic. These methanogenic bacteria can therefore compete with other bacterial groups such as sulphate-reducing bacteria.63 The SRB may also be involved in the hydrolysis64 and acetogenesis steps.65 In addition, the SRB are known to play a key role in the biodegradation of a number of environmental pollutants.66
The strain LINDBL, isolated at 37°C (Table 4) in the presence of 20mM lactate, is most closely related to Desulfobulbus propionicus with a 99% sequence similarity. D. propionicus was first isolated from a fluidised bed bioreactor treating effluent containing acid and metals.67 Strains LINDBH and LINDBH2 were affiliated phylogenetically to Desulfovibrio vulgaris with 99% sequence similarity. Both strains were isolated at 37°C on basal medium supplemented with H2CO2 (2 bars) as a substrate. D. vulgaris is used as a model for the study of SRB to analyse the mechanisms of metal corrosion and to treat toxic metal ions from the environment.68
LINDBA1 and LINDBHT1 are two mesophilic strains that were isolated on basal medium for SRB using two different substrates, the first one in the presence of 20mM acetate and the second one in the presence of H2CO2 (at 2 bars). These strains have Desulfomicrobium baculatum as their closest relative, with 99% sequence similarity. Phylogenetic analysis demonstrated that the strain LINDBHT1T belonged within the genus Desulfotomaculum. This strain (LINDBHT1T) had Desulfotomaculum halophilum69 and Desulfotomaculum alkaliphilum70 as its closest phylogenetic relatives with approximately 89% sequence similarity. LINDBHT1T is a novel anaerobic thermophilic sulphate-reducing bacterium. This bacterium constitutes a new species of the genus Desulfotomaculum, D. peckii71 (Fig. 3).

Various SRB were isolated from the anaerobic digestor which shows that they are involved in the degradation process. In order to gain an overall idea of the cultivable bacterial diversity of the digestor, we grouped all of the bacteria isolated on to the same phylogenetic tree (Fig. 4).

Analysis of the microbial populations obtained from the anaerobic sludge samples in both mesophilic and thermophilic conditions led to the isolation of many morphologically distinct bacteria. Molecular and microbial analyses showed that fermentative bacteria (primarily Clostridium spp. and Parabacteroides spp.), Desulfobulbus spp., Desulfomicrobium spp., Desulfovibrio ssp. and Desulfotomaculum ssp. were the prominent members of the bacterial community in the bioreactor (Fig. 4). The diversity of the microbial community within the digestor may reflect the metabolic diversity of microorganisms involved in anaerobic digestion. The interactions of this complex microbial community allows for complete degradation of natural polymers such as polysaccharides, proteins, lipids and nucleic acids into methane and carbon dioxide.
ConclusionIn conclusion, the use of both bacterial culture and molecular techniques enabled us to establish a picture of the existing microbial biodiversity in an anaerobic digestor. The culture approach was essential, especially with regard to culture and/or isolation of microorganisms with no known cultivable representative. Further research in this area can only improve our knowledge of microbial anaerobic digestors, including the role of different microbial populations involved in anaerobic degradation of waste, which will improve control of these treatment processes. A comprehensive molecular inventory would also support and complement our study of the microbial diversity of anaerobic cultures as it would link information on the diversity and function of microbial communities in their environment.
Conflicts of interestThe authors have no conflict of interest to declare.