


Primary immunodeficiencies (PID) represent a heterogeneous group of genetic disorders characterised by poor or absent function in one or more components of the immune system. Humoral or antibody immunodeficiencies are the most common form of PID, of which common variable immunodeficiency (CVID) is the most frequent symptomatic form. CVID is usually characterised by hypogammaglobulinaemia with poor antibody specificity, and an increased susceptibility to infections, autoimmunity and lymphoproliferation. Fewer than 10% of CVID patients have a known monogenic basis. Several chromosomal abnormalities (chromosome 18q-syndrome, monosomy 22, trisomy 8 and trisomy 21) are currently identified as causes of hypogammaglobulinaemia, and can manifest with recurrent infections and mimic CVID.
MethodsReview of clinical charts and laboratory results of paediatric patients followed in the outpatient clinic of PID with a diagnosis of genetic disease and humoral immunodeficiency.
ResultsThree patients with different genetic diseases (19p13.3 deletion, a ring 18 chromosome and Kabuki syndrome), were identified. During follow-up, they developed signs and symptoms suggestive of humoral deficiency mimicking CVID, despite which immunoglobulin levels were quantified with considerable delay with respect to symptoms onset, and specific management was subsequently delayed.
ConclusionsPatients with genetic abnormalities and recurrent infections should be evaluated for hypogammaglobulinaemia. An early diagnosis of humoral deficiency can allow treatment optimisation to prevent complications and sequelae.
Primary immunodeficiencies (PID) represent a heterogeneous group of genetic disorders characterised by poor or absent function in one or more components of the immune system. Currently there are at least 250 genetically defined inborn errors of immunity, which are classified into nine categories by the International Union of Immunology Societies (IUIS), according to the part of the immune system that is most dysfunctional.
Humoral immunodeficiencies are the most common form of PID; they are characterised by alterations in the development and function of B lymphocytes resulting in a defect in the synthesis of antibodies, manifested by reduced or absent serum immunoglobulin levels with poor specificity, and an increased susceptibility to infections (mainly of the respiratory and gastrointestinal tract).1 Common variable immunodeficiency (CVID) is the most frequent from of symptomatic humoral deficiency and, besides infections, can also involve other manifestations such as autoimmunity and lymphoproliferation. The CVID diagnostic criteria comprise: hypogammaglobulinaemia with IgG levels two standard deviations below the mean for age and a marked decrease in at least one of the isotypes IgM or IgA; impaired vaccine responses or absent isohaemagglutinins; and exclusion of other causes of hypogammaglobulinaemia.2,3 There are several causes of hypogammaglobulinaemia, which should be considered in the differential, which include chromosomal abnormalities of which chromosome 18q-syndrome, monosomy 22, trisomy 8 and trisomy 21 are currently included in the guidelines.2,3 These patients can present with recurrent infections that are often ascribed to the chromosomal abnormality, and hypogammaglobulinaemia is usually not ruled out. The aim of this study was to describe three cases with different genetic diseases that during follow-up developed signs and symptoms suggestive of humoral deficiency, despite which immunoglobulin levels were quantified with considerable delay with respect to symptoms onset.
Materials and methodsOf the 18 paediatric patients followed in the Immunodeficiency Outpatient Clinic at Sant Joan de Déu Hospital in Barcelona with a diagnosis of CVID-like humoral deficiency (based on hypogammaglobulinaemia with IgG levels two standard deviations below the mean for age and a marked decrease in at least one of the isotypes IgM or IgA; impaired vaccine responses or absent isohaemagglutinins,2,3 and symptoms (recurrent sinopulmonary infections, autoimmune and lymphoproliferative diseases)), we identified three children who were diagnosed with a genetic disease, and during follow-up had developed signs and symptoms suggestive of humoral deficiency. Immune evaluation of these patients included: immunoglobulin levels (IgG, IgA, IgM), IgG subclasses levels if >4 years old, T/B/NK lymphocytes phenotyping, antibodies against polysaccharide antigens (isohaemagglutinins and pneumococcus, post-infection), and protein antigens (diphtheria, tetanus, pneumococcus post-Prevenar vaccination), B cell subphenotyping and proliferative responses to mitogens, using standard methods.1 No statistical methods were used in this study.
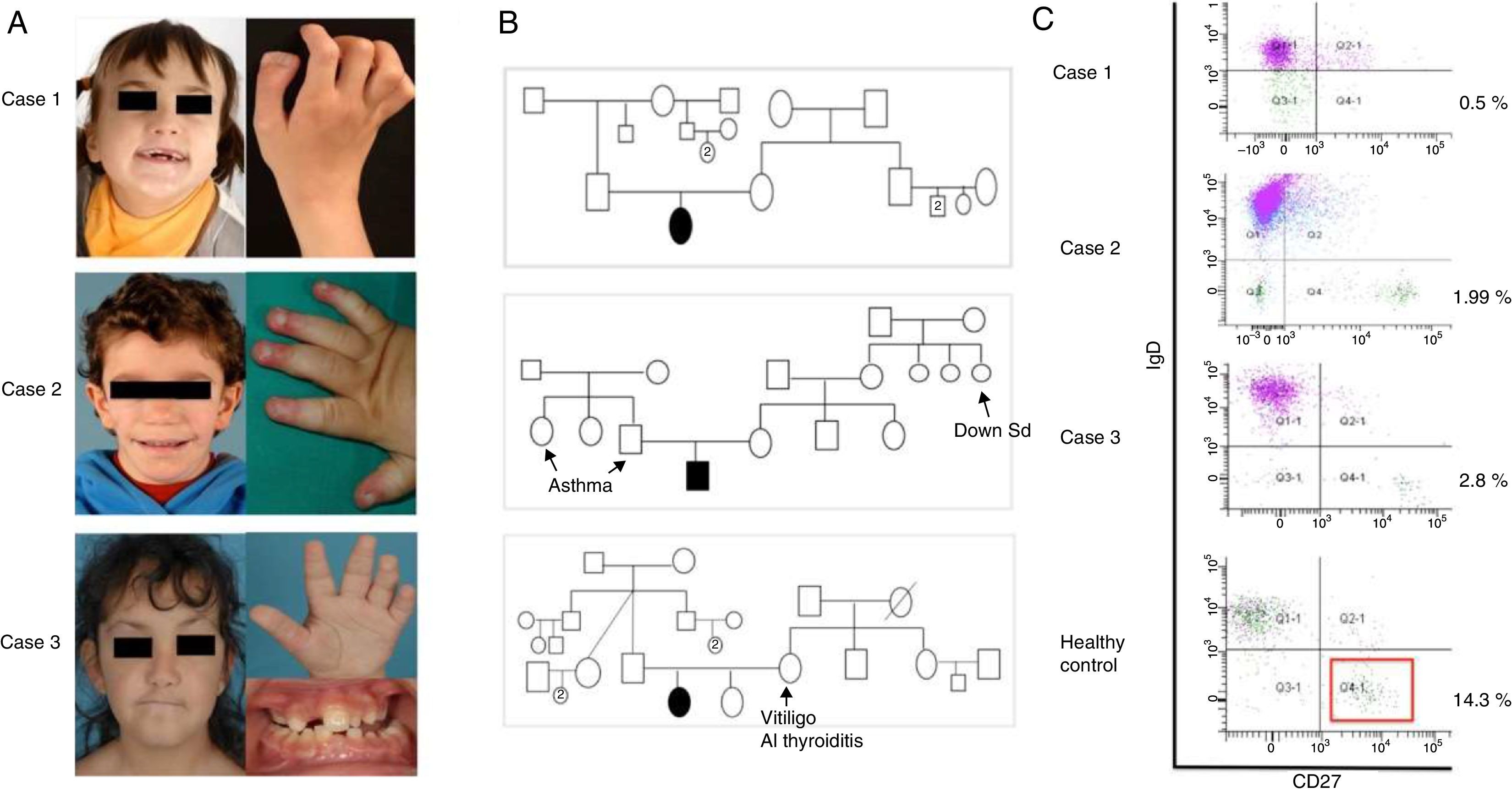
ResultsThe first case is a seven-year-old female born to healthy unrelated parents at 37 GA by C-section due to delayed in utero growth (birth weight 2.02kg). Screening during pregnancy showed broad nuchal fold and enlargement of the lateral ventricles with normal foetal karyotype (46 XX). In the neonatal period, she manifested irritability, weak suction and dysmorphic features (Fig. 1). Subtelomeric analysis by FISH technique showed a 19p13.3 deletion. Further evaluation identified an hypoplasia of the corpus callosum and ventriculomegaly, with psychomotor retardation, dysphagia and refractory epilepsy, treated with different combinations of drugs (clobazam and topiramate first, followed by phenobarbital, ethosuximide and valproic acid); limb abnormalities (duplication of the distal phalanx of the right thumb and genu varum); and endocrinopathy (growth retardation, hypothyroidism and adrenal insufficiency, all under replacement since age five). At six years old, she started presenting recurrent sinopulmonary infections, including recurrent suppurative otitis media, and five episodes of bronchopneumonia, one of which required hospitalisation for respiratory distress (clinical data are summarised in Table 1). Immunological investigations (Table 2) were conducted at seven years of age, showing low IgG, IgA and IgG subclasses (IgG1, IgG2 and IgG4) with normal responses to vaccines, including Pneumovax®23, but with progressive loss of protective anti-pneumococcal levels eight months after vaccination. She is being evaluated for the dysphagia as a main trigger of respiratory infections, along with hypogammaglobulinaemia. She has not initiated immunoglobulin replacement yet, pending clinical and immunological evolution.
 Case 1 (19p13.13), facies at seven years old, and duplication of the distal phalanx of the right thumb. Case 2 (ring 18), facies at five years old, and acrodermatitis in distal phalange. Case 3 (Kabuki), facies at seven years old, and persistence of foetal fingertip pads and dental anomalies and malocclusion. (B) Family trees, (C) Flow cytometry: decreased switched-memory B cells (IgD-CD27+) compared to age-matched control.")
Clinical data collected from three affected patients. (A) Case 1 (19p13.13), facies at seven years old, and duplication of the distal phalanx of the right thumb. Case 2 (ring 18), facies at five years old, and acrodermatitis in distal phalange. Case 3 (Kabuki), facies at seven years old, and persistence of foetal fingertip pads and dental anomalies and malocclusion. (B) Family trees, (C) Flow cytometry: decreased switched-memory B cells (IgD-CD27+) compared to age-matched control.
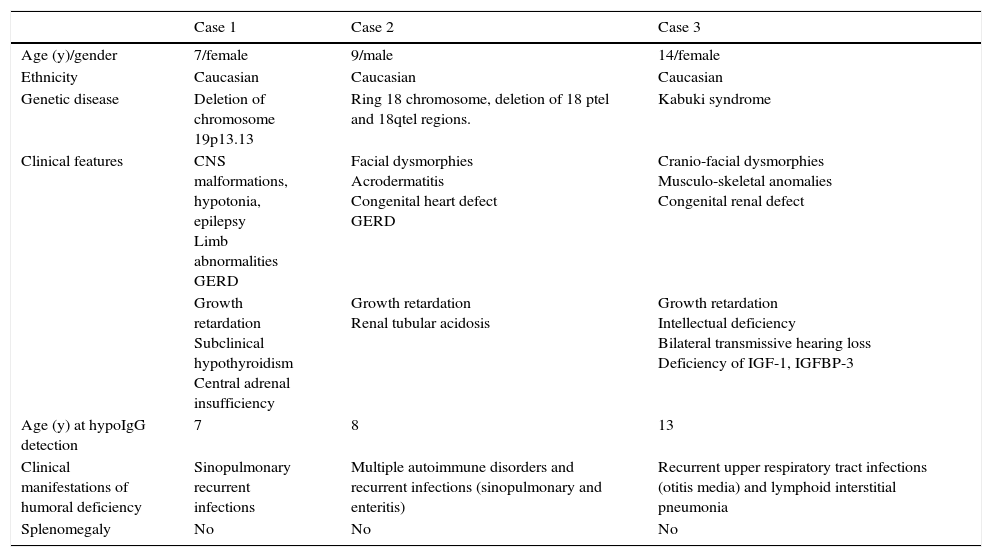
Clinical characteristics of patients with genetic disease and humoral immunodeficiency.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age (y)/gender | 7/female | 9/male | 14/female |
| Ethnicity | Caucasian | Caucasian | Caucasian |
| Genetic disease | Deletion of chromosome 19p13.13 | Ring 18 chromosome, deletion of 18 ptel and 18qtel regions. | Kabuki syndrome |
| Clinical features | CNS malformations, hypotonia, epilepsy Limb abnormalities GERD | Facial dysmorphies Acrodermatitis Congenital heart defect GERD | Cranio-facial dysmorphies Musculo-skeletal anomalies Congenital renal defect |
| Growth retardation Subclinical hypothyroidism Central adrenal insufficiency | Growth retardation Renal tubular acidosis | Growth retardation Intellectual deficiency Bilateral transmissive hearing loss Deficiency of IGF-1, IGFBP-3 | |
| Age (y) at hypoIgG detection | 7 | 8 | 13 |
| Clinical manifestations of humoral deficiency | Sinopulmonary recurrent infections | Multiple autoimmune disorders and recurrent infections (sinopulmonary and enteritis) | Recurrent upper respiratory tract infections (otitis media) and lymphoid interstitial pneumonia |
| Splenomegaly | No | No | No |
Abbreviations: GERD, gastro-oesophageal reflux disease.
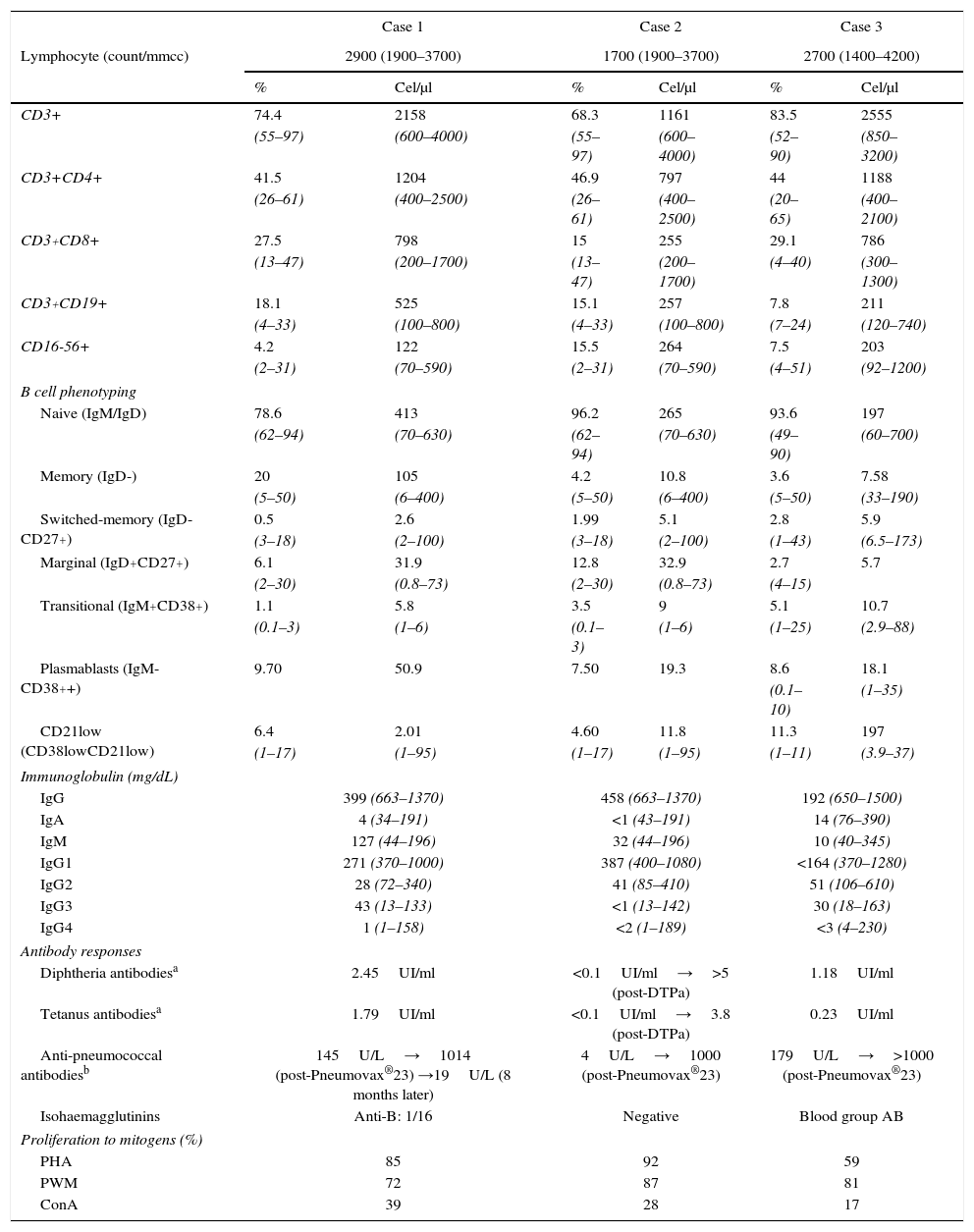
Immune characteristics of patients with genetic disease and humoral immunodeficiency.
| Case 1 | Case 2 | Case 3 | ||||
|---|---|---|---|---|---|---|
| Lymphocyte (count/mmcc) | 2900 (1900–3700) | 1700 (1900–3700) | 2700 (1400–4200) | |||
| % | Cel/μl | % | Cel/μl | % | Cel/μl | |
| CD3+ | 74.4 | 2158 | 68.3 | 1161 | 83.5 | 2555 |
| (55–97) | (600–4000) | (55–97) | (600–4000) | (52–90) | (850–3200) | |
| CD3+CD4+ | 41.5 | 1204 | 46.9 | 797 | 44 | 1188 |
| (26–61) | (400–2500) | (26–61) | (400–2500) | (20–65) | (400–2100) | |
| CD3+CD8+ | 27.5 | 798 | 15 | 255 | 29.1 | 786 |
| (13–47) | (200–1700) | (13–47) | (200–1700) | (4–40) | (300–1300) | |
| CD3+CD19+ | 18.1 | 525 | 15.1 | 257 | 7.8 | 211 |
| (4–33) | (100–800) | (4–33) | (100–800) | (7–24) | (120–740) | |
| CD16-56+ | 4.2 | 122 | 15.5 | 264 | 7.5 | 203 |
| (2–31) | (70–590) | (2–31) | (70–590) | (4–51) | (92–1200) | |
| B cell phenotyping | ||||||
| Naive (IgM/IgD) | 78.6 | 413 | 96.2 | 265 | 93.6 | 197 |
| (62–94) | (70–630) | (62–94) | (70–630) | (49–90) | (60–700) | |
| Memory (IgD-) | 20 | 105 | 4.2 | 10.8 | 3.6 | 7.58 |
| (5–50) | (6–400) | (5–50) | (6–400) | (5–50) | (33–190) | |
| Switched-memory (IgD-CD27+) | 0.5 | 2.6 | 1.99 | 5.1 | 2.8 | 5.9 |
| (3–18) | (2–100) | (3–18) | (2–100) | (1–43) | (6.5–173) | |
| Marginal (IgD+CD27+) | 6.1 | 31.9 | 12.8 | 32.9 | 2.7 | 5.7 |
| (2–30) | (0.8–73) | (2–30) | (0.8–73) | (4–15) | ||
| Transitional (IgM+CD38+) | 1.1 | 5.8 | 3.5 | 9 | 5.1 | 10.7 |
| (0.1–3) | (1–6) | (0.1–3) | (1–6) | (1–25) | (2.9–88) | |
| Plasmablasts (IgM-CD38++) | 9.70 | 50.9 | 7.50 | 19.3 | 8.6 | 18.1 |
| (0.1–10) | (1–35) | |||||
| CD21low (CD38lowCD21low) | 6.4 | 2.01 | 4.60 | 11.8 | 11.3 | 197 |
| (1–17) | (1–95) | (1–17) | (1–95) | (1–11) | (3.9–37) | |
| Immunoglobulin (mg/dL) | ||||||
| IgG | 399 (663–1370) | 458 (663–1370) | 192 (650–1500) | |||
| IgA | 4 (34–191) | <1 (43–191) | 14 (76–390) | |||
| IgM | 127 (44–196) | 32 (44–196) | 10 (40–345) | |||
| IgG1 | 271 (370–1000) | 387 (400–1080) | <164 (370–1280) | |||
| IgG2 | 28 (72–340) | 41 (85–410) | 51 (106–610) | |||
| IgG3 | 43 (13–133) | <1 (13–142) | 30 (18–163) | |||
| IgG4 | 1 (1–158) | <2 (1–189) | <3 (4–230) | |||
| Antibody responses | ||||||
| Diphtheria antibodiesa | 2.45UI/ml | <0.1UI/ml→>5 (post-DTPa) | 1.18UI/ml | |||
| Tetanus antibodiesa | 1.79UI/ml | <0.1UI/ml→3.8 (post-DTPa) | 0.23UI/ml | |||
| Anti-pneumococcal antibodiesb | 145U/L→1014 (post-Pneumovax®23) →19U/L (8 months later) | 4U/L→1000 (post-Pneumovax®23) | 179U/L→>1000 (post-Pneumovax®23) | |||
| Isohaemagglutinins | Anti-B: 1/16 | Negative | Blood group AB | |||
| Proliferation to mitogens (%) | ||||||
| PHA | 85 | 92 | 59 | |||
| PWM | 72 | 87 | 81 | |||
| ConA | 39 | 28 | 17 | |||
Reference values: Schatorjé EJ, Gemen EF, Driessen GJ, et al. Age-matched reference values for b-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. 2011;74(5):502–10.
The second case is a nine-year-old male born at term (41 weeks GA) with adequate weight for GA, from healthy unrelated parents. From birth he had mild hypotonia and dysmorphic features with acrodermatitis in distal phalange (Fig. 1). He was diagnosed with renal tubular acidosis, congenital heart defect (membranous subaortic stenosis), gastro-oesophageal reflux disease (GERD), mild psychomotor retardation and growth hormone deficiency (under replacement). A conventional chromosome GTG-banding at a 550 band level was performed, showing a ring 18 chromosome, with no other structural or numerical anomalies; more than 50 metaphases were revised and no mosaicism was found. The genetic study was completed with subtelomeric regions analysis by FISH technique: the results showed deletion of 18ptel and 18qtel regions. During the first two years of life he presented several infections: omphalitis one month of age, three non-suppurative otitis media, recurrent bronchitis, and a multilobar pneumonia for which he was admitted to the paediatric intensive care unit. Initially, the immunological study showed only low IgA levels. Over time he developed recurrent pneumonia, recurrent diarrhoea with positive stool culture for Giardia lamblia, multiple autoimmune disorders (vitiligo, autoimmune hypothyroidism (on replacement therapy), hepatitis with positive anti-smooth muscle antibodies, positive parietal cells antibodies, severe panniculitis with extensive lipoatrophy, fasciitis and myositis and recurrent episodes of urticaria without trigger) (Table 1). At eight years old, his immune status was re-evaluated and low levels of IgG, IgM and IgG subclasses (IgG1, IgG2, IgG3) were detected in addition to the low IgA levels (Table 2). Antibodies against diphtheria and tetanus were negative after completing the regular calendar schedule, but turned positive after a booster dose. Baseline pneumococcal antibodies after recurrent infections of supposed pneumococcal origin were negative, but turned positive after a dose of Pneumovax®23 (Table 2). He was started on intravenous immunoglobulin replacement with good clinical and laboratory response of both infectious and autoimmune manifestations. CT scan shows no bronchiectasis.
The third case is a 14-year-old female born at 38 GA by C-section, with low birth weight per gestational age (2.50kg). She has healthy unrelated parents and a healthy eight-year-old sister. She has craniofacial dysmorphic features (coronal craniosynostosis, low-set ears, arched and sparse eyebrows, long palpebral fissures with eversion of the lower lids, dental anomalies and malocclusion, Fig. 1), intellectual deficiency with full scale IQ of 52, musculo-skeletal anomalies (scoliosis, congenital dislocation of both hips, valgus feet, femur patellar instability of both knees), horseshoe kidney, dermatoglyphic abnormalities (persistence of foetal fingertip pads), and intrauterus growth retardation without catch-up, deficit of IGF-1, IGFBP-3, under hormonal replacement (Table 1). These findings are consistent with Kabuki syndrome (KS). The MLPA study found no duplications or deletions in MLL2 (KMT2D) gene, nor in KDM6A gene. She started manifesting recurrent infections at 10 years of age, characterised by recurrent otitis media 1–2/month, and urinary tract infections with positive urine culture to Escherichia coli. For this reason at 13 years of age an immunological study was conducted showing extremely low IgG, IgA, IgM and IgG subclasses (IgG1, IgG2, IgG4) with positive vaccine responses to diphtheria and tetanus after completing the regular calendar schedule, but low pneumococcal antibodies despite recurrent middle ear infections, which turned positive after a dose of Pneumovax®23 (Table 2). CT scan was compatible with lymphoid interstitial pneumonia. She was started on intravenous immunoglobulin replacement followed by subcutaneous immunoglobulin, with good clinical and laboratory evolution. The T/B/NK lymphocyte phenotyping and the study of proliferation to mitogens (Con A, PHA, and PWM) were normal in all three cases. B cell subphenotyping study showed alterations (reduction in memory B cells and/or switched-memory B cells) in all three cases (Table 2).
DiscussionCommon variable immune deficiency (CVID) is a heterogeneous group of primary humoral deficiencies with hypogammaglobulinaemia, in which the molecular basis is still unclear. During the last 10 years, 10–15% of the patients with CVID phenotype have been reclassified after identifying specific gene defects in ICOS, CD19, CD20, CD81, CD21, BAFFR, LRBA, PRKCD, NFKB2, IL21 (OMIM # 607594).4 Herein we report three patients with a genetic disease, in whom the clinical and laboratory findings overlap with CVID: all three cases had recurrent sinopulmonary infections associated with hypogammaglobulinaemia and variable impairment of vaccine responses, along with B cell sub-phenotyping alterations (reduction in memory B cells and/or switched-memory B cells typically found in CVID patients).1 Case 2 had first low levels of IgA followed over the years by low levels of IgG and IgM, as described in some cases of CVID. Besides, two patients developed autoimmune and/or immune dysregulation complications: case 2 (ring chromosome 18) had multiple autoimmune disorders and case 3 (KS) presented CT scan images compatible with lymphoid interstitial pneumonia, complications similarly described in patients with CVID.5 None had splenomegaly.
In KS, recurrent infections, especially otitis media and pneumonia, are present in approximately 60% of patients,6 but it is unclear if this condition is secondary to immune dysfunction. One study on 19 children with KS revealed 79% had low IgA levels and 42% also had low IgG levels.7 Lymphoid interstitial pneumonia in KS has been previously described.8 In chromosome 19 defect, there is one previously reported case of a boy with cryptic subtelomeric deletion of 19p13.3-pter that had recurrent upper respiratory tract infections with immune dysfunction (low IgA, IgG2, and low pneumococcal antibodies); other cases with similar features, but with different chromosomal 19 aberration (ring chromosome 19 and 19p13.12 deletion) have been described, but none reported an immune dysfunction in their findings.9 Alterations in immunoglobulin levels have been described in patients with short arm deletion (18p-), long arm deletion (18q-), and ring formation (18r), especially low or absent levels of IgA, which have evolved to a humoral immunodeficiency compatible with CVID (18q-),6 similar to our observation in case 2 (ring 18 chromosome with deletion of 18ptel and 18 qtel regions).
There are other causes of secondary hypogammaglobulinaemia such as drugs, antiepileptic drugs in particular,2 which must be taken into account in patients with genetic disorders, since they often suffer from epilepsy. Indeed, carbamazepine, phenytoin and valproic acid have been described to induce a secondary B cell defect.10 In case 1, the patient was treated with valproic acid but this treatment was started two months after the observation of hypogammaglobulinaemia of IgG and IgA, which dismisses it as the cause of her hypogammaglobulinaemia. Interestingly, all three patients were receiving treatment with GH, although there are no reports of a relationship between the use of GH and hypogammaglobulinaemia.
The interest of these three observations is that they reinforce the theory of a genetic basis of CVID, at least in some cases, probably polygenic with the accumulation in different errors in genes involved in B cell maturation that leads to the final phenotype during early adulthood. In this sense, identifying genetic diseases that manifest with a CVID-like phenotype is of upmost interest, because it identifies gene locus that might be associated with CVID. Moreover, this report highlights the fact that a number of syndromes with single gene or chromosomal abnormalities associated with humoral immunodeficiency are not currently included in the group of known genetic causes associated with hypogammaglobulinaemia, such as Kabuki syndrome and chromosome 19 deletions. Indeed, the identification of these patients at risk of developing CVID-like disease is of upmost relevance for early diagnosis of humoral deficiency, which can allow treatment optimisation to prevent complications and sequelae.
In conclusion patients with genetic abnormalities and recurrent infections should be evaluated for hypogammaglobulinaemia. An early diagnosis of humoral deficiency can allow treatment optimisation to prevent complications and sequelae.
Sources of support and fundingThis study was supported by Fondo de Investigacion Sanitaria (FIS, project number PI12/01990 to LA).
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on the patients for this investigation. The data and sample collection were performed in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
We thank the patients and their families for their collaboration in this study.
