












El síndrome de Sjögren es una enfermedad autoinmunitaria sistémica con un alto impacto individual y social. El compromiso pulmonar presenta múltiples manifestaciones, con impacto en calidad de vida y riesgo de mortalidad. El abordaje dinámico integrado mediante un grupo de diagnóstico multidisciplinario que incluya expertos en neumología, reumatología, radiología y patología tiene el potencial de impactar en la identificación, las estrategias de manejo y los desenlaces. Aunque es necesario reconocer tempranamente a los pacientes con mayor riesgo, en la actualidad no se cuenta con biomarcadores confiables. Las estrategias de manejo farmacológico se basan en la inmunomodulación, pero la evidencia para su uso es de baja calidad. Promover el entrenamiento y la sensibilización del personal de salud podría reducir los retrasos en el acceso a una evaluación especializada.
Sjögren's syndrome is a systemic autoimmune disease with a high burden for the individual, as well as society. Pulmonary compromise presents with a myriad of manifestations that influence patient quality of life and mortality risk. An integrated dynamic approach by a multidisciplinary diagnostic discussion team that includes experts in chest diseases, rheumatology, radiology, and pathology has the potential to improve the identification, management strategies, and outcomes. Although early recognition of patients at high risk is essential, there is currently a lack of reliable biomarkers. Pharmacological therapies are based on immunomodulation, although the evidence to support their use is of low quality. Increasing awareness and training among healthcare professionals may reduce a delayed access to specialized assessment.
El síndrome de Sjögren (SSj) es una enfermedad autoinmunitaria sistémica que se caracteriza por síntomas secos, los cuales suelen asociarse con dolor articular y fatiga, con un profundo impacto en la productividad de los pacientes1. Su prevalencia a escala mundial oscila entre el 0,01 y el 2,7%, compromete particularmente a las mujeres con una relación mujer: hombre que puede alcanzar 9:11,2.
Aunque se considera una exocrinopatía que afecta en mayor medida a las glándulas salivares y lacrimales, las manifestaciones sistémicas, dentro de las cuales se incluye el compromiso pulmonar, pueden encontrarse hasta en el 40% de los pacientes; existen diferentes estrategias clinimétricas que permiten evaluar los diferentes sistemas afectados1,3.
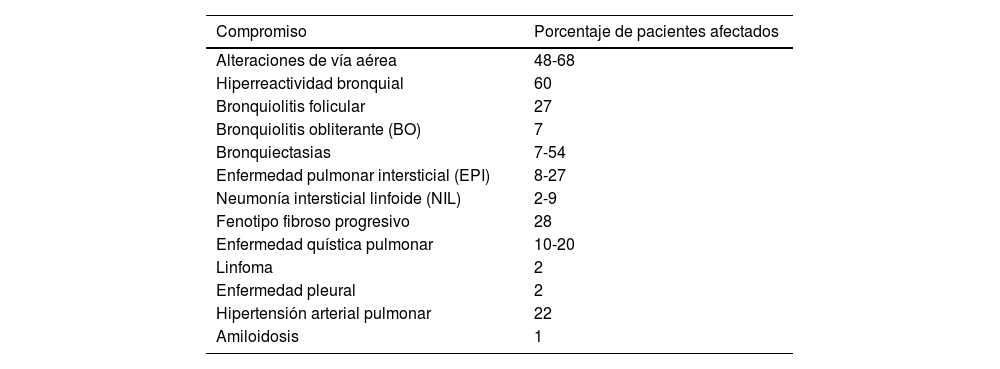
Los síntomas respiratorios en SSj son frecuentes, con prevalencias que oscilan entre el 9 y el 75%, según la definición utilizada y la población evaluada; la tos y la disnea pueden estar presentes hasta en el 50% de los pacientes4. Los estudios que definen el compromiso pulmonar como «la presencia de síntomas acompañados de anormalidades imagenológicas o en las pruebas de función pulmonar» estiman una prevalencia que supera el 20%5. Cerca del 10% de los pacientes puede iniciar con compromiso pulmonar6,7 y se ha reportado que hasta el 20% presenta compromiso pulmonar intersticial a los cinco años del diagnóstico de la enfermedad8. La frecuencia de este compromiso puede manifestarse de múltiples maneras, como se resume en la tabla 1 y en la figura 19-11.
Frecuencia de las manifestaciones pulmonares del síndrome de Sjögren
| Compromiso | Porcentaje de pacientes afectados |
|---|---|
| Alteraciones de vía aérea | 48-68 |
| Hiperreactividad bronquial | 60 |
| Bronquiolitis folicular | 27 |
| Bronquiolitis obliterante (BO) | 7 |
| Bronquiectasias | 7-54 |
| Enfermedad pulmonar intersticial (EPI) | 8-27 |
| Neumonía intersticial linfoide (NIL) | 2-9 |
| Fenotipo fibroso progresivo | 28 |
| Enfermedad quística pulmonar | 10-20 |
| Linfoma | 2 |
| Enfermedad pleural | 2 |
| Hipertensión arterial pulmonar | 22 |
| Amiloidosis | 1 |

En el 2013, la American Thoracic Society (ATS) y la European Respiratory Society (ERS) emitieron una actualización en el diagnóstico y la clasificación de las enfermedades pulmonares intersticiales (EPI), en la que se define como método de referencia para el diagnóstico el «abordaje dinámico integrado», generado a la luz de la discusión de un equipo multidisciplinario12. La evidencia reciente ha demostrado la existencia de barreras de acceso a estos abordajes en países de bajos y medianos ingresos, como Colombia13, por lo que una mayor sensibilización al personal de salud podría reducir los retrasos en el acceso a una evaluación especializada. Presentamos una revisión actualizada de las manifestaciones pulmonares en pacientes con SSj, así como su abordaje y tratamiento.
MétodosSe llevó a cabo una revisión narrativa no sistemática, mediante la búsqueda de literatura en cuatro bases de datos (Cochrane Database, Pubmed/Medline, Embase y LILACS [que incluye literatura latinoamericana]). Se filtraron por idioma y se incluyeron artículos en inglés, español, francés, italiano, portugués y alemán. La estrategia de búsqueda incluyó los siguientes términos (MeSH/DeCS o descriptores Explode): Sjögren's syndrome, lung, pulmo*, airway*. Se revisó también literatura gris usando estos términos en Google y se aplicó una estrategia de «bola de nieve» para recuperar la literatura útil de los artículos revisados. Los autores seleccionaron y revisaron los artículos basados en su contenido y relevancia.
¿Por qué son importantes las manifestaciones pulmonares en el síndrome de Sjögren?Múltiples estudios han reflejado su impacto en calidad de vida y mortalidad. Se ha descrito una menor calidad de vida en pacientes con compromiso pulmonar cuando se comparan con aquellos sin alteraciones14,15. También se ha descrito un mayor riesgo de mortalidad en pacientes con compromiso pulmonar, definido como la «presencia de síntomas y anormalidades en imágenes o pruebas de función pulmonar», que es hasta cuatro veces mayor que en pacientes sin compromiso pulmonar, con una supervivencia a 10 años de 73 vs. 92%15.
Lin et al. reportaron factores asociados con mortalidad, entre ellos el compromiso pulmonar intersticial (Odds Ratio, OR 3,2, 1,2-8,6) y la hipertensión pulmonar (OR 6,4, 2-20,1), a pesar de intervalos de confianza amplios16. Nannini et al. encontraron resultados similares, sin alcanzar significación estadística8. Estos datos resaltan la importancia de identificar a aquellos pacientes con mayor riesgo de desarrollar compromiso pulmonar. Hasta el momento algunos factores se han asociado con un mayor riesgo: duración más prolongada de la enfermedad, edad avanzada, sexo masculino y tabaquismo; se han reportado algunas características serológicas que serán abordadas más adelante5.
Manifestaciones pulmonares del síndrome de SjögrenXerotráquea y xerobronquitisLa afectación de la vía aérea es la manifestación más frecuente. Es causada por la destrucción de las glándulas exocrinas y por infiltración de linfocitos T CD4+ en la submucosa bronquial o bronquiolar. El compromiso de la tráquea y de los bronquios se manifiesta como sequedad de la mucosa (i. e., xerotráquea y xerobronquitis) y se caracteriza predominantemente por tos seca; puede asociarse con una alteración funcional obstructiva9,10.
Se ha encontrado que la depuración mucociliar se halla significativamente disminuida, comparada con la de los controles (3,3 ± 1,2 mm/min vs. 5,9 ± 1,1 mm/min), y en aquellos que tienen xerotráquea la depuración es indetectable. Como consecuencia, las secreciones viscosas se acumulan y se impactan produciendo atelectasias, bronquiectasias y episodios recurrentes de bronquitis y neumonía17.
Enfermedades de la vía aérea pequeñaLa enfermedad de la vía aérea pequeña es la alteración pulmonar más frecuente en pacientes sintomáticos respiratorios. La infiltración linfocítica difusa de la vía aérea se ha relacionado con tos crónica, hiperreactividad bronquial (reportada hasta en el 60% de los pacientes) y bronquitis crónica18-20. Sus manifestaciones radiológicas incluyen bronquiectasias y bronquiolitis (fig. 2). Los pacientes con SSj tienen un mayor riesgo de desarrollar asma (Hazard Ratio, HR 1,38, 1,21-1,58) y enfermedad pulmonar obstructiva crónica (HR ajustado 1,9, 1,1-1,75)21-24.

Varios estudios han descrito cambios funcionales consistentes con anormalidades bronquiolares, incluso en pacientes sin síntomas respiratorios25. En una serie de 14 pacientes con SSj llevados a biopsia, se encontró bronquiolitis folicular en el 27% de ellos, bronquiolitis crónica en el 21% y bronquiolitis obliterante (BO) en el 7%26,27. Desde el punto de vista radiológico, la bronquiolitis se caracteriza por nódulos centrilobulillares, con densidad de vidrio esmerilado, mal definidos y árbol en gemación28. La principal causa de estas alteraciones puede relacionarse con compromiso infeccioso. Sin embargo, en aquellos pacientes sin clínica de infección o con este diagnóstico razonablemente descartado, tales hallazgos pueden ser la manifestación de una bronquiolitis folicular.
Bronquiolitis folicularEsta forma de bronquiolitis es la más frecuente en el SSj y se puede manifestar clínicamente con disnea sibilante. Los hallazgos imagenológicos son variables: en una serie de 12 pacientes con bronquiolitis folicular confirmada por biopsia, la tomografía computarizada de tórax de alta resolución (TACAR) mostró con más frecuencia opacidades reticulares, nódulos pequeños y opacidades en vidrio esmerilado26,29. El pronóstico es bueno, los pacientes usualmente responden al manejo con glucocorticoides o en algunos casos con el empleo de macrólidos26.
Cuando el compromiso se limita al folículo linfoide peribronquiolar, la enfermedad se manifiesta como bronquiolitis folicular. Cuando el infiltrado compromete los septos alveolares, el espectro abarca la neumonía intersticial linfoide (NIL), que se abordará más adelante11.
Bronquiolitis obliteranteLa BO o bronquiolitis constrictiva es una entidad caracterizada por la presencia de fibrosis de los bronquiolos terminales y distales, así como hallazgos de obstrucción en la espirometría que suelen ser progresivos30. En una serie publicada por Wight et al., la incidencia de BO fue del 36%, que, en la mayoría de los casos, mostró alivio tras el tratamiento con glucocorticoides, cloroquina o ciclofosfamida31.
BronquiectasiasLa prevalencia de bronquiectasias diagnosticadas por TACAR, en SSj, se encuentra entre el 7 y el 54%32. En una serie de 54 pacientes con SSj y bronquiectasias, todas fueron cilíndricas y el 71% se localizó en los lóbulos inferiores; los autoanticuerpos anti-Ro/SSA estuvieron presentes con menor frecuencia en los pacientes con bronquiectasias. Durante el seguimiento se encontró que el 56% de los pacientes presentó neumonía, siendo más frecuente que en los pacientes sin bronquiectasias33,34.
Enfermedad pulmonar intersticialEl compromiso pulmonar intersticial en el SSj puede manifestarse con diferentes patrones histológicos y radiológicos. Los principales aspectos de la enfermedad pulmonar intersticial (EPI), asociada con SSj, fueron analizados en una revisión sistemática reciente de 20 estudios realizada por Sambataro et al. La EPI ocurre en 8-27% de los pacientes con SSj. Se presenta en el año siguiente al diagnóstico en el 10% de ellos, cinco años después en el 20% y a los 15 años en el 47%; no obstante, entre el 10 y el 50% de los casos puede presentarse años antes de la aparición de los síntomas de SSj8,35. Las cohortes han reportado una edad media de 55 a 61 años, con una relación hombre:mujer de 2:8 y una incidencia en fumadores de alrededor del 20%.
Los síntomas respiratorios como tos y disnea se reportan en 40-66% de los pacientes y pueden estar relacionados con xerotráquea o con la presencia de enfermedad obstructiva; entre el 24 y el 38% de los pacientes con EPI no informa síntomas respiratorios8,35. Sin embargo, la tos seca, la edad avanzada, el tabaquismo, el hipocratismo digital, la pérdida de peso y un focus score ≥ 4, en la biopsia de glándula salival menor, se encuentran con mayor frecuencia en los pacientes con SSj y EPI. Así mismo, se ha reportado la alta incidencia de reflujo gastroesofágico (50%) y niveles más altos de IgM, deshidrogenasa láctica, velocidad de eritrosedimentación globular, proteína C reactiva y positividad para anti-Ro/SSA8,35,36.
En los estudios en los que se ha realizado biopsia pulmonar, en su mayoría quirúrgica, el patrón de neumonía intersticinal no específica (NINE) es el más común, particularmente del subtipo fibrótico, en lugar del celular. La frecuencia del patrón de neumonía intersticial usual (NIU) aumenta hasta un 33%, y la amiloidosis, así como el linfoma, pueden verse hasta en el 10%6,35. Aunque tradicionalmente la biopsia quirúrgica ha sido utilizada como procedimiento de referencia para el estudio de las EPI, recientemente se ha introducido la criobiopsia como una alternativa menos invasiva y en la que es posible aproximarse a los patrones histológicos de las EPI en el contexto de enfermedades del tejido conectivo37.
En cuanto a los hallazgos en la TACAR, todos los estudios coinciden en que el patrón de EPI más común es el de NINE, que ocurre en el 41-45% de los pacientes, seguido por el patrón de NIU en aproximadamente el 10%, neumonía en organización en el 4% y NIL en el 4-9%; hasta en el 40% de los sujetos puede verse una combinación de estos patrones35,38,39. El patrón de NINE se caracteriza por opacidades en vidrio esmerilado. Si bien el gradiente ápico-basal no es una característica determinante para su diagnóstico, la gran mayoría de los pacientes exhibe mayor afectación de los lóbulos inferiores y hasta en el 30% de los casos puede no haber afectación subpleural basal. La presencia de opacidades reticulares y bronquiectasias o bronquiolectasias de tracción puede relacionarse con el patrón fibrótico de esta entidad clínica (fig. 3)28.
. Vidrio esmerilado en lóbulo medio, língula y lóbulos inferiores con bronquiectasias de tracción. No hay evidencia de panal de abejas.")
En relación con los hallazgos histopatológicos del patrón de NINE, los cambios típicos son difusos: se observa un compromiso homogéneo del parénquima pulmonar de tipo celular (fig. 4) o fibrótico (fig. 5), sin presencia de áreas de parénquima conservado40.
 Hematoxilina eosina: expansión intersticial difusa y homogénea, predominantemente celular, consistente con una neumonía intersticial no específica (NINE celular).")
 Hematoxilina eosina: compromiso intersticial de predominio fibrótico difuso con escasa celularidad, consistente con una neumonía intersticial no específica (NINE).")
Por su parte, aunque los cambios radiológicos típicos del patrón de NIU son difusos y se caracterizan por la presencia de panal de abejas con una distribución basal y subpleural, no todos los casos de NIU confirmados con biopsia presentan este patrón radiológico28. El patrón de NIU de los pacientes con SSj suele diferenciarse de las formas idiopáticas (i. e., fibrosis pulmonar idiopática) (fig. 6A-B); esto además se relaciona con la edad de presentación en los pacientes con SSj que suelen ser mayores y predominantemente mujeres. Las adenomegalias mediastinales pueden verse con frecuencia41. Sus hallazgos histopatológicos se caracterizan por fibrosis parcheada, predominantemente subpleural y paraseptal, con áreas de parénquima pulmonar conservado, focos fibroblásticos, panalización y presencia de agregados y folículos linfoides en el parénquima pulmonar sin fibrosis12. Los pacientes con patrón de NIU y SSj tienden a tener mejor respuesta a la inmunomodulación y mejor pronóstico que los pacientes con fibrosis pulmonar idiopática41.
 Extenso panal de abejas de predominio basal con bronquiectasias de tracción. En los lóbulos superiores se identifican lesiones cavitadas de pared gruesa e irregular, la de mayor tamaño en el lado derecho con material de tejidos blandos en su interior. En este paciente, con antecedente de SSj, se concluyó patrón de neumonía intersticial usual (NIU) y se comprobó aspergilosis crónica cavitada asociada.")
A-B) Extenso panal de abejas de predominio basal con bronquiectasias de tracción. En los lóbulos superiores se identifican lesiones cavitadas de pared gruesa e irregular, la de mayor tamaño en el lado derecho con material de tejidos blandos en su interior. En este paciente, con antecedente de SSj, se concluyó patrón de neumonía intersticial usual (NIU) y se comprobó aspergilosis crónica cavitada asociada.
Para determinar la extensión del compromiso radiológico se han propuesto puntajes visuales que permiten hacer una evaluación semicuantitativa de la TACAR, aun con una alta variabilidad inter- e intraobservador38,42. Recientemente se ha reportado la utilidad de los índices cuantitativos en SSj, que son obtenidos a partir de software especializado43.
Con relación al compromiso funcional, la mayoría de los estudios reporta una reducción en la capacidad de difusión del monóxido de carbono (DLCO), entre 37 y 54% del predicho con preservación de la capacidad vital forzada (CVF, valor medio de 72 a 82%). Se cree que esta diferencia entre la reducción de la DLCO y la estabilidad de la CVF puede explicarse porque el proceso inflamatorio afecta la membrana alveolar, lo que reduce la DLCO. La disminución de la CVF se presenta posteriormente, cuando la fibrosis ya está avanzada o por hipertensión pulmonar; esta última puede encontrarse hasta en el 12% de los pacientes35,44.
En los pacientes con EPI y SSj se ha reportado una supervivencia del 84% a cinco años, a pesar de un riesgo de mortalidad mayor comparado con pacientes sin compromiso pulmonar. Las causas más frecuentes de muerte son la exacerbación aguda y el compromiso infeccioso pulmonar. Dentro de los factores asociados con mortalidad se encuentran un volumen espiratorio forzado en el primer segundo (VEF1) y una CVF menores del 60%, hipercapnia en los gases arteriales, presencia de panal de abejas y reticulación extensa en la TACAR, así como hallazgo de focos fibroblásticos en la biopsia pulmonar16,35.
No existen estudios clínicos aleatorizados que evalúen el impacto del tratamiento de la EPI en SSj. La combinación más frecuente de tratamiento incluye prednisona oral 0,5-1 mg/kg día, más ciclofosfamida 100 mg/día por nueve meses, con titulación del glucocorticoide a lo largo de varios meses hasta una dosis de 5-7,5 mg/día; otras opciones de manejo incluyen azatioprina o hidroxicloroquina35,45.
Neumonía en organizaciónEs una manifestación poco frecuente en SSj que se caracteriza por síntomas subagudos con grados variables de tos y disnea. Las exacerbaciones agudas pueden semejar neumonías infecciosas. Sus hallazgos radiológicos son principalmente consolidaciones migratorias en patrones variados como subpleural, peribronquial y en banda, q con opacidades en vidrio esmerilado. La presencia de opacidades perilobulares o en halo reverso (i. e., signo del atolón) pueden evidenciar el diagnóstico12,28. Su principal característica histopatológica son tapones fibroblásticos que ocupan los espacios alveolares con relativa preservación del intersticio46.
Otros tipos de enfermedad pulmonar intersticialLa EPI asociada con SSj también puede ocurrir con una presentación aguda. Se han descrito casos de neumonía intersticial aguda y de neumonía en organización aguda fibrinosa (AFOP, acute fibrinous organizative pneumoniae), con una respuesta aceptable al empleo de glucocorticoides sistémicos47-51. Otra presentación de EPI descrita es la de fibroelastosis pleuroparenquimatosa, una EPI rara que se caracteriza por proliferación intersticial y subpleural de fibras elásticas en lóbulos superiores y engrosamiento pleural prominente; se ha reportado asociada con enfermedades autoinmunitarias y en una serie reciente de SSj se documentó en el 29% de los casos. La entidad con frecuencia es progresiva y fatal52-54.
¿Qué es el fenotipo fibroso progresivo de enfermedad pulmonar intersticial?Una proporción de pacientes con EPI asociada con enfermedades autoinmunitarias sistémicas desarrollan una forma progresiva fibrosante, caracterizada por aumento de la fibrosis en la TACAR, incremento de disnea, deterioro de la calidad de vida, empeoramiento de la función pulmonar, desarrollo de exacerbaciones y aumento de la mortalidad temprana55,56. Se ignora la proporción de pacientes con EPI autoinmunitaria que desarrollan fibrosis progresiva; sin embargo, una encuesta aplicada a reumatólogos y neumólogos con experiencia en el manejo de pacientes con EPI estimó que afectaba del 24 al 31% de los pacientes55-57. Al combinar la información de esta encuesta con una revisión sistemática de la literatura especializada58,59 se ha estimado que 13-40% de los pacientes con EPI autoinmunitaria desarrollan un fenotipo fibrosante progresivo. En SSj dos estudios han reportado falta de eficacia del tratamiento en el 28% de los pacientes en quienes se documentó progresión de la EPI35,59.
La identificación de este fenotipo es importante a la luz de los resultados del estudio INBUILD, en el que se evaluó la efectividad del nintedanib, un inhibidor de tirosina-cinasa que ha mostrado reducción en la tasa de progresión de la EPI en diferentes grupos de pacientes60. En este ensayo doble ciego, controlado con placebo, realizado en 15 países, se asignaron al azar 663 pacientes con EPI fibrosante de diversas etiologías, y que afectaba a más del 10% del volumen pulmonar en la TACAR, a recibir nintedanib en dosis de 150 mg dos veces al día o placebo. Todos los pacientes cumplieron criterios para progresión de la EPI en los últimos 24 meses, a pesar del tratamiento, y tenían una CVF de al menos el 45% del valor predicho y una DLCO entre 30 y 80% del valor predicho. En la población general la tasa ajustada de disminución de la CVF fue de -80,8 mL por año con nintedanib y -187,8 mL por año con placebo, para una diferencia entre grupos de 107 mL por año (65,4-148,5; p < 0,001).
En pacientes con un patrón fibrótico tipo NIU, la tasa ajustada de disminución en la CVF fue de -82,9 mL por año con nintedanib y -211,1 mL por año con placebo, para una diferencia de 128,2 mL (70,8-185,6; p < 0,001). La diarrea fue el evento adverso más común, como se informó en 66,9 y 23,9% de los pacientes tratados con nintedanib y placebo, respectivamente60.
De los 663 pacientes del estudio, 171 (25,8%) tenían EPI relacionada con enfermedades autoinmunitarias, de las cuales las más comunes fueron artritis reumatoide (n = 88), esclerosis sistémica (n = 40) y enfermedad mixta del tejido conectivo (n = 20); casi las tres cuartas partes de los pacientes con EPI autoinmunitaria (74,3%) tenían un patrón fibrótico tipo NIU en la TACAR61,62. Los hallazgos de un análisis de subgrupos mostraron que en estos pacientes el medicamento redujo en 58% la tasa anual de declinación de la CVF, con una diferencia de 102,7 mL/año (IC 95% 23,2-182,2; p = 0,012) en comparación con placebo61,62.
De esta forma, se ha sugerido que en todo paciente con EPI asociada con autoinmunidad, una vez establecido el diagnóstico e iniciado el tratamiento específico, se debe llevar a cabo un seguimiento periódico con el fin de identificar dicho fenotipo progresivo y considerar tratamiento antifibrótico (fig. 7)63.

Algoritmo de evaluación recomendado para pacientes con EPI asociada con autoinmunidad, con el fin de identificar los casos de fenotipo fibroso progresivo. Modificado de Cottin.63.
Cuando los pacientes con SSj presentan compromiso pulmonar, la presencia de quistes (p. ej., lesiones de espacio aéreo de paredes de menos de 2 mm) es característica y puede representar un patrón de NIL, bronquitis folicular, hiperplasia folicular linfoide, NINE, amiloidosis o linfoma de tejido linfoide asociado con las mucosas (mucosa-associated lymphoid tissue, MALT)64,65. El desarrollo de los quistes puede deberse a dos mecanismos: 1) el efecto de válvula con dilatación y 2) la destrucción del espacio alveolar e infiltración linfoide. Con frecuencia se presentan en los lóbulos inferiores o con distribución peribroncovascular; se acompañan de opacidades en vidrio esmerilado y nódulos66,67. La presencia de quistes o enfermedad pulmonar en estos pacientes supedita un aumento en mortalidad y disminución de la calidad de vida15.
Neumonía intersticial linfoideLa NIL es una EPI rara que se encuentra dentro del espectro de alteraciones linfoproliferativas no neoplásicas y que tiene estrecha relación con enfermedades autoinmunitarias. Frecuentemente se asocia con infecciones y enfermedad del tejido conectivo (más del 80% de los casos) y su forma idiopática es rara, por lo que en el Consenso de Neumonías Intersticiales Idiopáticas del año 2013, de ATS/ERS, pasó a ser parte del grupo de enfermedades intersticiales raras12. Hasta un 50% de los pacientes que la presentan tiene SSj y los demás casos se relacionan con artritis reumatoide, lupus eritematoso sistémico (LES), polimiositis, inmunodeficiencia común variable y virus de la inmunodeficiencia humana (VIH)68-70.
Su frecuencia es mayor en mujeres y representa el 15% de los casos de EPI en SSj. Se caracteriza por hiperplasia del tejido linfoide asociado con el bronquio (bronchus-associated lymphoid tissue, BALT) que ocasiona infiltración difusa de los septos alveolares por linfocitos policlonales68. Sus síntomas más comunes son disnea, tos y pérdida de peso. Al examen físico se pueden encontrar estertores velcro a la auscultación pulmonar e hipocratismo digital71-73. Los hallazgos en la radiografía de tórax son variables y dependen de la severidad de la enfermedad. Las manifestaciones pueden ir desde una radiografía normal hasta un aumento en el tamaño de los pulmones con visualización de lesiones quísticas y opacidades reticulares finas relacionadas con la pared de las lesiones quísticas.
Ante la sospecha de NIL, el estudio de elección es la TACAR. La presencia de quistes de distribución peribroncovascular se evidencia hasta en el 68% de los casos y se identifica hasta en el 46% de los pacientes con SSj74,75. Los quistes son de distribución aleatoria y su tamaño puede ser variable, en un rango entre los 3 mm y los 5 cm (fig. 8A-C). Otros hallazgos descritos son patrón en vidrio esmerilado, nódulos centrilobulillares pobremente definidos y engrosamiento del intersticio con patrón linfangítico74. Los hallazgos histopatológicos se caracterizan por un componente inflamatorio linfoplasmocitario muy denso que se acompaña de formación de numerosos folículos linfoides, los cuales distorsionan la arquitectura pulmonar, en la que se debe hacer diagnóstico diferencial con linfomas de bajo grado46.
 Patrón de neumonía intersticial linfoide (NIL). Tomografía computarizada de tórax de alta resolución que evidencia el característico patrón de quistes de pared fina. B-C) Paciente con diagnóstico de infección por virus de la inmunodeficiencia humana (VIH), sintomático respiratorio. B) Radiografía de tórax: pulmones aumentados de tamaño con patrón micronodular fino de predominio central y basal. C) Tomografía computarizada de tórax de alta resolución: corte axial. Micronódulos centrilobulillares. Lavado bronquio-alveolar negativo para infección. Biopsia compatible con neumonía intersticial linfoide (NIL).")
A) Patrón de neumonía intersticial linfoide (NIL). Tomografía computarizada de tórax de alta resolución que evidencia el característico patrón de quistes de pared fina. B-C) Paciente con diagnóstico de infección por virus de la inmunodeficiencia humana (VIH), sintomático respiratorio. B) Radiografía de tórax: pulmones aumentados de tamaño con patrón micronodular fino de predominio central y basal. C) Tomografía computarizada de tórax de alta resolución: corte axial. Micronódulos centrilobulillares. Lavado bronquio-alveolar negativo para infección. Biopsia compatible con neumonía intersticial linfoide (NIL).
Algunos casos de NIL pueden progresar a un patrón fibrótico con panal de abejas. En caso de diagnóstico diferencial con linfoma, teniendo claro que la NIL no progresa hacia este, se debe realizar biopsia pulmonar para establecer el diagnóstico definitivo por inmunohistoquímica: si se trata de NIL demostrará policlonalidad en el tejido pulmonar, mientras que en caso de ser cáncer mostrará monoclonalidad76.
Honda et al. encontraron que los quistes estaban presentes en el 82% de pacientes con NIL vs. solo el 2% en pacientes con linfoma, lo que sugiere que si bien el linfoma es un diagnóstico diferencial, tal presentación es mucho menos frecuente en este grupo77. La neumonía por Pneumocystis jirovecii es un diagnóstico diferencial, especialmente en pacientes con VIH. El tratamiento debe enfocarse en el control de la causa de base; el uso de glucocorticoides se asocia con buena respuesta clínica68.
LinfomaDentro del grupo de las enfermedades autoinmunitarias, el SSj presenta la mayor predisposición al desarrollo de linfomas, especialmente de tipo no Hodgkin78, con pocos casos de linfoma Hodgkin79. En los pacientes con linfoma es frecuente el compromiso extranodal; los linfomas MALT son los más comunes, con una prevalencia de 2% y hasta 44 veces más frecuentes en SSj que en la población general80,81. La oncogénesis del linfoma en estos pacientes se relaciona con la estimulación antigénica crónica que conlleva la selección monoclonal de linfocitos B y la posterior aparición de mutaciones80,81. Los sitios más frecuentemente afectados son las glándulas salivares, el cuello y el pulmón82. Los síntomas más frecuentes incluyen disnea, tos, síntomas B y ronquera, con una relación temporal de 2 a 15 años entre el diagnóstico de SSj y la aparición del linfoma83. Los predictores para el desarrollo de linfoma en pacientes con SSj incluyen linfopenia CD4+, hipocomplementemia, púrpura, crecimiento parotídeo, esplenomegalia, linfadenopatia y paraproteinemia84-86.
Los hallazgos radiológicos pulmonares en linfoma MALT incluyen nódulos solitarios o múltiples con áreas de consolidación o atenuación en vidrio esmerilado, bronquiectasias y engrosamiento septal en relación con el espacio broncovascular. También puede haber derrame pleural65-77.
Una vez realizado el diagnóstico, y con el tratamiento apropiado, el pronóstico es favorable, con supervivencia superior a 5 o 10 años en más del 90% de los pacientes, aunque puede variar según el tipo de linfoma80,84.
Hiperplasia nodular linfoideTambién denominada seudolinfoma pulmonar, es una proliferación linfoide benigna asociada con SSj. Su aparición se correlaciona con la severidad de los síntomas secos y puede progresar a linfoma. Se manifiesta de diferentes maneras, entre ellas como un nódulo o masa solitaria, múltiples nódulos, opacidades en vidrio esmerilado o consolidaciones múltiples en el pulmón. Su diagnóstico requiere biopsia para descartar compromiso por linfoma o neumonía en organización87. La histología muestra infiltrados de linfocitos T y B circunscritos con centros germinales y la inmunohistoquímica revela proliferación policlonal. La resección quirúrgica es curativa y se ha reportado buena respuesta al uso de glucocorticoides88.
Enfermedad pleuralEl compromiso pleural en pacientes con SSj es raro, con cohortes que describen una frecuencia menor al 1%. Las manifestaciones pleurales descritas comprenden líneas subpleurales, engrosamiento o derrame pleural. Estos hallazgos son en su mayoría asintomáticos89,90. Cuando hay derrame pleural, uni- o bilateral, la sintomatología más frecuente está dada por fiebre, tos, dolor pleurítico y disnea89,91. Al documentarse el derrame se requiere un estudio sistemático con toracentesis para descartar infección (bacteriana o por micobacterias) o incluso la presencia de linfoma, especialmente si se acompaña de adenopatías o nódulos pulmonares mayores a 1 cm91-93.
El derrame pleural primario por SSj es, característicamente, un exudado linfocitario con infiltración linfocitaria en las biopsias pleurales; los anticuerpos antinucleares (ANA) y antiRo/SSA pueden ser positivos en el líquido. En las diferentes series, el tratamiento del derrame incluye observación, pues puede resolverse de forma espontánea, o manejo con prednisolona en dosis de 30 a 60 mg/día, y en algunos casos pulsos de metilprednisolona, con buena respuesta clínica91,94.
Hipertensión pulmonar y tromboembolia pulmonarLa hipertensión arterial pulmonar (HAP) es mucho menos común en SSj que en otras enfermedades del tejido conjuntivo95-99. En una cohorte británica de 484 pacientes, con HAP asociada a enfermedades autoinmunitarias, solo tres pacientes tenían SSj primario, en comparación con 315 con esclerosis sistémica, 36 con enfermedad mixta del tejido conectivo, 35 con LES y 18 con artritis reumatoide99. En un estudio de 107 pacientes con SSj se documentó HAP por criterios ecocardiográficos en 24 (22%), pero sin correlación clínica100.
Con respecto a la supervivencia, en una cohorte china de 190 pacientes con HAP y enfermedades del tejido conjuntivo, esta fue superior en SSj (64,8%) comparada con la de pacientes con LES (61%) o esclerosis sistémica (43,9%) a los 5 años98. En otra cohorte de 29 pacientes con SSj y HAP (documentado por cateterismo derecho), las tasas de supervivencia a 1, 3 y 5 años fueron 80, 75 y 67%, respectivamente; el uso de la terapia inmunosupresora se asoció con mayor supervivencia (HR 0,11, 0,02-0,45; p = 0,004)97.
En cuanto al tratamiento, en un estudio retrospectivo de 30 pacientes japoneses con HAP y enfermedad del tejido conjuntivo, entre los que se encontraban pacientes con SSj, fueron tratados con inmunosupresores en monoterapia o combinados con vasodilatadores pulmonares. A corto plazo, las respuestas (definidas como mejora en la clase funcional de la Organización Mundial de la Salud [OMS]) se documentaron en 16 pacientes (53%).
El uso de inmunomoduladores, especialmente ciclofosfamida intravenosa y glucocorticoides, fue un factor de predicción independiente de respuesta a corto plazo. Las tasas acumuladas de muerte relacionada con HAP fueron menores en respondedores a corto plazo (p = 0,04) y en pacientes con diagnóstico concomitante de HAP y enfermedad del tejido conectivo tratados con inmunosupresores y vasodilatadores pulmonares101.
Dada la infrecuente presentación de la HAP en SSj, el tratamiento con prostanoides sistémicos, antagonistas de los receptores de endotelina e inhibidores de la fosfodiesterasa se extrapola de los estudios de HAP95,102-104.
Por otra parte, debe tenerse en cuenta que el SSj es un factor de riesgo independiente de enfermedad tromboembólica venosa. En una cohorte canadiense se reportó una incidencia de 3,9 casos por cada 1.000 personas-año de tromboembolia pulmonar y de 2,8 casos por cada 1.000 personas-año de trombosis venosa profunda, en comparación con cifras de 0,9 y 0,8 en la población general, hallazgos que fueron replicados en un estudio poblacional taiwanés105,106.
Amiloidosis pulmonarLa amiloidosis pulmonar relacionada con SSj es rara (2% de los pacientes con infiltrados) y cuando ocurre lo hace con depósito de cadenas ligeras AL (lambda o kappa) o, menos frecuentemente, amiloide AA, sin compromiso sistémico107,108. En una revisión de 37 casos de amiloidosis pulmonar en SSj, 96% de los casos se presentaron en mujeres con síntomas de tos crónica, disnea y dolor torácico. En las imágenes se observaron nódulos difusos sólidos o en vidrio esmerilado, con o sin calcificaciones, quistes de predominio en lóbulos inferiores, engrosamiento septal y consolidaciones. La biopsia pulmonar, requerida para establecer el diagnóstico y excluir linfoma, mostró infiltración de linfocitos y plasmocitos, así como depósitos de amiloide identificados con coloración de rojo Congo67,108.
Anticuerpos y compromiso pulmonarLa utilidad de los autoanticuerpos como factores asociados con el desarrollo de compromiso pulmonar en SSj es objeto de debate. La positividad de los ANA, descrita con mayor frecuencia en pacientes con compromiso pulmonar, ha llegado hasta el 70% de los pacientes36,109,110. Sin embargo, un estudio holandés mostró una relación inversa111 y un estudio turco no encontró relación112. La asociación con la presencia de factor reumatoide también es incierta y su rendimiento diagnóstico en un estudio con 30 pacientes con enfermedad pulmonar fue muy discreto, con valores predictivos positivo y negativo cercanos a 60%109-113.
Cerca de dos tercios de los pacientes con SSj son positivos para antiRo/SSA y hasta un tercio es positivo para antiLa/SSB36,110-114; aún no se ha encontrado asociación con el desarrollo de enfermedad pulmonar36,110-114. Por su parte, un estudio coreano no encontró asociación entre la positividad de antiRo y la mortalidad en pacientes con enfermedades reumáticas115. Sin embargo, recientemente Buvry et al. publicaron un estudio en el que reportaron que la presencia de anticuerpos antiRo52 se comporta como un factor de riesgo independiente para EPI en SSj116, en línea con lo descrito por Davidson et al. en el año 2000117.
La relevancia de los datos de Buvry et al. radica en que es el primer reporte que soporta la asociación de la presencia de antiRo52 y compromiso pulmonar en SSj. Esta asociación se había descrito previamente en numerosas enfermedades del tejido conectivo e incluso se ha relacionado con un peor pronóstico vital118-123. El antígeno SSA cuenta con dos proteínas conocidas como Ro52 (52 kDa) y Ro60 (60 kDa), que se asocian con sistemas diferentes de autoanticuerpos (i. e., son codificados por genes diferentes, cumplen funciones diferentes, se encuentran en compartimientos celulares diferentes e incluso son identificados por [auto]anticuerpos diferentes)124; los ensayos utilizados para la detección de Ro/SSA suelen utilizar una mezcla de ambas proteínas y hasta el 20% de los resultados negativos resulta positivo al evaluar las proteínas por separado, por lo que se ha propuesto su medición individual124.
Por otra parte, en una cohorte francesa de 467 pacientes con SSj se exploró el impacto de la positividad para antiRNP. Se encontraron 19 pacientes positivos, de los cuales solo tres cumplieron criterios para enfermedad mixta del tejido conjuntivo. Estos pacientes mostraron una mayor frecuencia de compromiso pulmonar (19 vs. 6%) y, llamativamente, de positividad para antiRo (90 vs. 67%)125.
TratamientoEl tratamiento del compromiso pulmonar en SSj dependerá del tipo y la severidad del compromiso (fig. 9). En la actualidad se encuentran en la literatura especializada las recomendaciones de manejo de diferentes asociaciones (en orden cronológico, iniciando desde la más reciente): European League Against Rheumatism 2020 (EULAR)126, British Society for Rheumatology 2017127, Japan College of Rheumatology 2017128, Institute of Medicine (IOM)/American Dental Association/American Academy of Ophthalmology (AAO)/American College of Rheumatology 2016129 y Sociedade Brasileira de Reumatologia 2015130. En general, la evidencia sobre el tratamiento es baja. No hay recomendaciones respecto a tamizaje de enfermedad pulmonar.

El tratamiento sistémico debe ser ajustado a la severidad del compromiso de órgano específico y las terapias sistémicas se deben limitar a pacientes con enfermedad sistémica, dada su pobre eficacia en síntomas glandulares126-128. La guía europea recomienda establecer la severidad del compromiso sistémico utilizando el instrumento ESSDAI (del inglés, EULAR Sjögren's Syndrome Disease Activity Index), que también permite evaluar la respuesta al tratamiento. Se define como respuesta a una mejoría de al menos tres puntos en el ESSDAI126. Sin embargo, no incluye la hipertensión pulmonar ni la pleuritis, por lo que deben ser individualizadas.
Las guías europeas y brasileña dividen el compromiso en dos grupos: bronquial e intersticial. Se sugiere el manejo del compromiso bronquial utilizando inhaladores (incluyendo glucocorticoides inhalados) y asociarlos con glucocorticoides sistémicos cuando sea necesario. En caso de no haber respuesta se debe considerar uso de inmunomoduladores como los planteados para compromiso intersticial; siempre se debe contar con la opinión de un experto126,130.
Para el compromiso intersticial, la guía europea propone establecer la severidad del compromiso usando ESSDAI. En caso de compromiso moderado o severo, los glucocorticoides sistémicos a dosis de 0,5-1 mg/kg/día son la primera línea de tratamiento. Esta estrategia se debe aplicar particularmente a NIL o a neumonía en organización; la efectividad en NINE y NIU es menor. Se debe procurar asociar un ahorrador de glucocorticoides (p. ej., inmunomodulador). La guía brasileña apoya esta postura, mientras que la británica sugiere asociarlo únicamente si no hay respuesta. Los inmunomoduladores sugeridos son azatioprina, micofenolato de mofetilo, metotrexato, ciclosporina y ciclofosfamida. La guía británica emite abiertamente una recomendación en contra del uso de leflunomida.
El uso de antimaláricos se menciona, pero no como primera opción de tratamiento. No existe evidencia que sugiera un inmunomodulador sobre otro, aunque parece existir más experiencia con el uso de azatioprina. La guía brasileña sugiere, en caso de no haber respuesta a glucocorticoides e inmunomodulador oral, el uso de metilprednisolona intravenosa más ciclofosfamida. Llamativamente, la guía japonesa describe que no existe suficiente evidencia que soporte la utilidad de los glucocorticoides en compromiso sistémico, más allá del osteomuscular o el cutáneo, incluyendo el pulmonar. El uso de glucocorticoides debe ser a las dosis más bajas posibles y por el menor tiempo posible. La guía americana no emite una recomendación particular respecto al manejo del compromiso pulmonar.
En casos refractarios, asociados con alta actividad de la enfermedad, debe considerarse inicialmente el uso de ciclofosfamida como terapia de rescate; todas las guías sugieren el uso de biológicos. Un revisión sistemática del grupo Cochrane, de la efectividad de la ciclofosfamida en EPI, mostró una mejoría en la CVF cuando se comparó contra placebo, sin encontrar diferencia al compararla con micofenolato mofetilo (MMF); los estudios revisados incluyeron en su mayoría pacientes con esclerosis sistémica, lo que limita la extrapolación a SSj131. Respecto de la terapia biológica, rituximab es la principal opción, seguido de belimumab y abatacept, aunque la guía británica no encuentra evidencia suficiente para recomendar los dos últimos. El uso de inmunoglobulinas puede ser considerado en casos de difícil manejo. Las guías europea, británica y japonesa emiten recomendaciones en contra del uso de antiTNF, particularmente infliximab. La guía británica no apoya el uso de anakinra o tocilizumab.
La guía brasileña emite una recomendación específica sobre evitar el tratamiento de las secuelas de la enfermedad fibrótica pulmonar.
Las guías disponibles no abordan el uso de antifibróticos (nintedanib, pirfenidona) en estos pacientes. Así mismo, no emiten recomendaciones específicas respecto al manejo de la hipertensión pulmonar.
ConclusiónEl SSj es la segunda enfermedad autoinmunitaria más frecuente a escala mundial y tiene un amplio rango de complicaciones sistémicas, entre las que destaca el compromiso pulmonar por su frecuencia, múltiples manifestaciones e impacto en calidad de vida y riesgo de mortalidad. La enfermedad pulmonar intersticial es probablemente la manifestación más severa y requiere el abordaje de un grupo de diagnóstico multidisciplinario para su identificación y manejo. Los instrumentos de clinimetría disponibles solo incluyen el compromiso intersticial, dejando de lado manifestaciones potencialmente severas como la hipertensión pulmonar. En la actualidad no se cuenta con biomarcadores confiables que permitan identificar tempranamente a los pacientes con mayor riesgo. Las estrategias de manejo farmacológico se basan en la inmunomodulación, sin embargo, la evidencia para su uso es de baja calidad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
